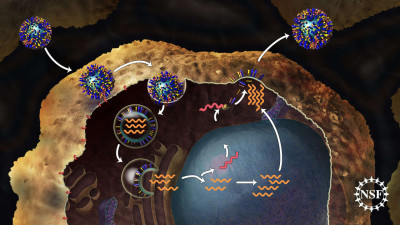
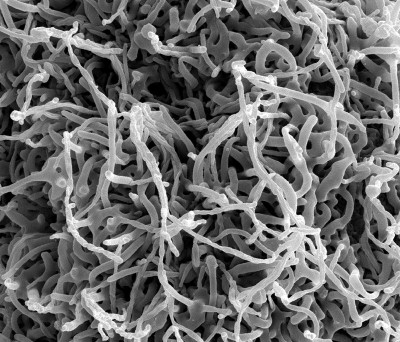
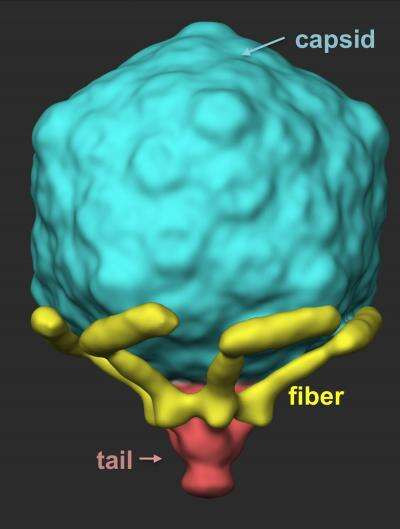
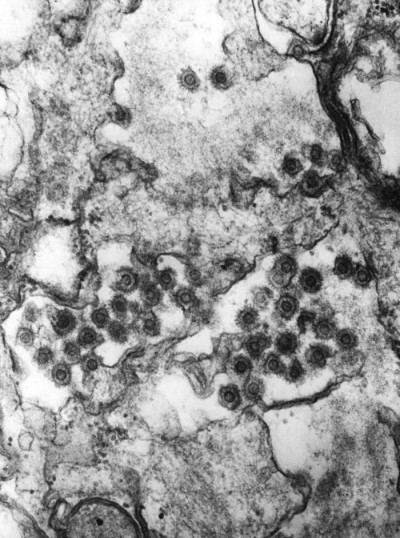
Image Gallery: An Exhibition of Scientific Fascination
Subject:
All Disciplines
Arts & Humanities
Biology & Life Sciences
Business & Economics
Chemistry & Materials Science
Computer Science & Mathematics
Engineering
Environmental & Earth Sciences
Medicine & Pharmacology
Physical Sciences
Public Health & Healthcare
Social Sciences