 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio Galvao | + 6393 word(s) | 6393 | 2021-04-27 11:53:00 | | | |
| 2 | Antonio Galvao | -3391 word(s) | 3002 | 2021-05-18 22:10:27 | | | | |
| 3 | Nora Tang | -1030 word(s) | 1972 | 2021-05-19 08:38:40 | | |
Video Upload Options
Obesity is a prevalent disease worldwide, usually associated with infertility. Studies in obese infertile females show the occurrence of systemic hyperestrogenemia, hyperinsulinemia, and associated ovarian dysfunction through premature follicular atresia and anovulation. Indeed, the ovaries of obese mothers have been shown to accumulate lipids, high levels of reactive oxygen species, and inflammatory mediators. Furthermore, obesity has been shown to hamper not only oocyte maturation and quality but also embryo development, with reported long term effects and direct causality between obesity in the mother and prevalence of cardiovascular disease or cancer in the offspring.
1. Introduction
The worldwide epidemic of obesity has reached unprecedented levels and infertility is described as an associated comorbidity [1]. Indeed, obese women were linked to poor reproductive outcomes, such as anovulation or decreased conception rate [2]. Literature has evidenced the link between peripheral insulin resistance and functional hyperandrogenism and hyperestrogenism [3], the main causes of anovulation and reduced endometrial receptivity [1]. Studies in mice have shown how obesity-associated hyperinsulinemia and diabetes can lead to delayed oocyte maturation in preovulatory follicles and apoptosis in granulosa cells (GC) [4]. Furthermore, ovaries of obese women were shown to present high levels of androstenedione and testosterone [3], responsible for premature follicular atresia. Therefore, the endocrine imbalance observed in obese mothers affects directly the follicular pool, reducing the number of primordial follicles and compromising fertility [5].
2. Leptin—A Common Denominator between Ovarian Function and Obesity
During obesity progression, the ever-growing adipose tissue secretes large amounts of leptin, which causes systemic hormonal imbalance. Leptin is mainly known to regulate appetite at the central level [6] besides modulating the release of gonadotropin releasing hormone (GnRH) neuron activity and gonadotropins [7]. Nonetheless, leptin is also an important modulator of ovarian function [8]. Leptin long and short receptor isoforms were detected in most cell types in murine ovary, particularly in the oocyte [8]. Likewise, both isoforms of the leptin receptor (ObR) were previously detected in human GC and theca cells (TC) [9]; as well, leptin and leptin soluble receptor were detected in human follicular fluid [9][10]. Thus, leptin signaling components’ heavy representation in the ovary makes the organ particularly vulnerable to the systemic hyperleptinemia observed in obese mothers [11].
Concerning the role of leptin in the ovary, evidence gathered from studies with leptin or ObR deficient mice, as well as women, confirmed their infertility and altered pubertal development [12][13]. Functionally, leptin actions in the ovary were shown to have a bimodal nature. In vivo and in vitro studies in mouse ovarian explants [14] evidenced the dose-dependent effect of leptin on progesterone (P4) synthesis, with low doses stimulating and high doses inhibiting expression of enzymes involved in P4 synthesis [14]. Also, studies in other species corroborated the aforementioned observations as the in vitro treatment of equine luteal cells with lower doses of leptin supported P4 secretion, whereas higher doses presented no effect [15]. Particularly regarding follicular dynamics, studies in mice showed that high levels of circulating leptin blocked folliculogenesis, but lower circulating levels of leptin supported the transition from primary to secondary follicle [16]. Conversely, the in vitro treatment of mouse follicles with mouse recombinant leptin once more revealed a dose-dependent response with higher treatment doses inhibiting follicular growth [17]. Presently, we used the data from Zhang and co-workers’ recent report profiling the transcriptome of GC and oocytes isolated from different stages of folliculogenesis in women [18] and plotted the main components of the leptin signaling pathway in order to understand if their expression profile could be linked to a leptin concerted role during particular developmental stages. Indeed, we confirmed some leptin signaling components, such as protein tyrosine phosphatase non-receptor type 2 (PTPN2), protein tyrosine phosphatase (PTP) 1B, or STAT3, were abundantly transcribed in both oocytes and GCs throughout folliculogenesis, whereas others, such as SOCS3, presented very low expression in both cell types under physiological conditions, particularly in later stages of folliculogenesis (Figure 1). Finally, we have recently shown that obesity progression in DIO mice alters leptin signaling in the ovary with increased leptin signaling in the ovary of 4 wk DIO mice, being followed by the establishment of leptin resistance in the ovaries of 16 wk DIO mice [19]. Indeed, expression levels of SOCS3 in ovarian extracts were dramatically increased already at 4 wk DIO [19]. Therefore, our findings, as well as the observations on leptin signaling component expression in oocytes and GCs from women’s follicles, clearly suggest that the impairment of leptin signaling in the ovaries of obese mothers may contribute to pathogenesis of ovarian failure, particularly given leptin’s established role in ovarian function.
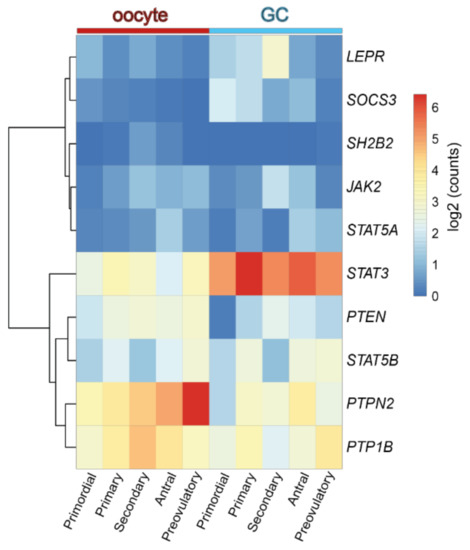
Figure 1. Heatmap representing the expression level of transcripts from the leptin signaling pathway components in human oocyte and granulosa cells (GC) throughout folliculogenesis. Primordial = primordial follicle; Primary = primary follicle; Secondary = secondary follicle; Antral = antral follicle; Preovulatory = preovulatory follicle. Color code from blue to red indicates the relative gene expression level from low to high, respectively. Data from Zhang et al. 2018 [18]. Leptin receptor (LEPR), suppressor of cytokine signaling 3 (SOCS3), SH2B Adaptor Protein 1 (SH2B1), Janus kinase 2 (JAK2), signal transducer and activator of transcription (STAT), protein tyrosine phosphatase non-receptor type 2 (PTPN2), protein tyrosine phosphatase (PTP) 1B.
3. Ovarian Function in Obese Mothers
The complex nature of folliculogenesis regulation certainly accounts for the vulnerability the ovaries present to the hormonal imbalance seen in obese mothers [2]. Generally, obesity may affect the oocyte and somatic cells at each single developmental stage of folliculogenesis with the incidence of obesity earlier in life posing a greater threat for the quality of the gamete later in adulthood [20]. Hence, in this section, we revisit folliculogenesis, analyzing major cellular events taking place throughout the long journey the female gamete makes from primordial follicle until fertilization, based on lessons learnt from studies in mice and in humans.
3.1 Obesity and Primordial Follicle Activation
Studies in rodents have highlighted the effects of maternal obesity on ovarian failure and, particularly, on PI3K dysregulation. A report in rats showed that DIO treatment led to POF through activation of mTOR and suppression of sirtuin (SIRT) 1 signaling [21], whereas the ovaries of mice fed a high-fat diet (HFD) presented aberrant expression levels of PI3K pathway components [22]. Indeed, the putative crosstalk between leptin and the PI3K pathway can be anticipated, mainly, through components of the insulin signaling cascade (Figure 2). After ObRb activation, JAK2 can phosphorylate IRs, with subsequent activation of PI3K pathway [23]. This generates PIP3, which further activates PIP3-dependent serine/threonine kinases, such as PDK1,2, responsible for activation of Akt, as previously discussed (Figure 2) [23]. Indeed, a study in mice with a triple mutation in ObR tyrosines linked follicle loss to the activation of PTEN/PI3K/Akt/mTOR signaling [24]. Furthermore, Panwar and colleagues showed the direct effects of leptin on the follicular pool once passive immunization of prepubertal mice against leptin prompted the transition of primordial to primary follicles [16]. Therefore, adequate levels of leptin signaling in the ovary seem to prevent primordial follicle hyperactivation and help to maintain the follicular pool. Concordantly, these observations suggest the decrease in leptin signaling we have reported in the ovaries of 16 wk DIO mice [19] might accelerate the activation and depletion of the primordial follicle pool.
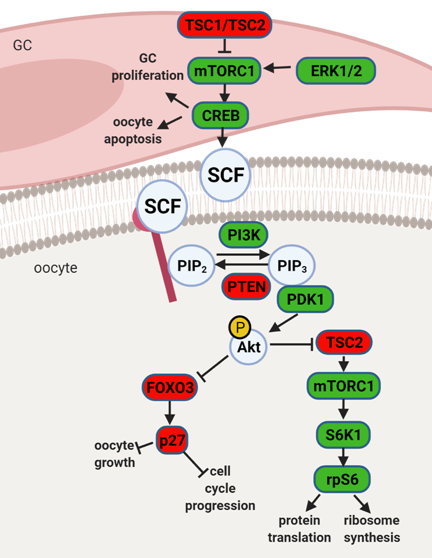
Figure 2. Schematic representation of mammalian target of rapamycin (mTOR) and phosphatidylinositol 3 kinase (PI3K) signaling pathway and its downstream regulators in the granulosa cells (GC) and oocyte. Extracellular signal-regulated protein kinase ½ (ERK1/2) activates mTOR complex 1 (mTORC1) in pre-GC to initiate the activation of primordial follicles. mTORC1 activates cyclic AMP-response element binding protein (CREB), which promotes stem cell factor (SCF) transcription and stimulates PI3K signaling but also affects pre-GC proliferation and oocyte apoptosis. mTOR signaling negative regulators TSC1 and TSC2 suppress mTORC1 activity. PI3K signaling pathway is activated by SCF in the oocyte. PI3K phosphorylates phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), which interacts with 3-phosphoinositide dependent protein kinase 1 (PDK1) for subsequent phosphorylation of protein kinase B (Akt) and forkhead box O3 (FOXO3) with its downstream mediator, cyclin-dependent kinase inhibitor 1B (p27). Upon phosphorylation, the inhibitory effect of FOXO3 and p27 on cell cycle progression and oocyte growth is inhibited, and the primordial follicle is recruited. Akt also activates ribosomal protein S6 kinase beta-1 (S6K1) through inhibition of TSC2 with subsequent mTORC1 activation and further ribosomal protein S6 (rpS6) phosphorylation, which leads to protein translation and ribosome synthesis. Factors indicated in red are associated with follicle dormancy; molecules indicated in green are associated with follicle activation. Created with BioRender.com, accessed on 1 November 2020.
3.2 Obesity and Preovulatory Follicle Formation
Obesity has been strongly associated with increased circulating levels of both E2 and androgens, not only due to the ability of the adipose tissue to synthesize steroids [25] but also to low circulating sex hormone binding globulin levels and suppression of gonadotropin release. Moreover, ovarian TC are known to respond to insulin during androgen synthesis [1]. Seminal work by Wu and co-workers clearly showed the link between hyperinsulinemia in DIO mice and increased ovarian androgen production through hyperactivation of CYP17 at TC level [26]. On the other hand, Souter et al. also correlated gain in body mass index (BMI) with E2 secretion by preovulatory follicles in obese women [27]. Finally, studies in rats evidenced the link between obesity and the lack of preovulatory surge of P4 and LH [28]. Hence, obesity clearly disrupts steroidogenesis, causing a predisposition to premature follicular atresia and anovulation.
3.3 Oocyte Maturation and Early Embryo Development in Obese Mothers
Obesity leads to severe systemic hormonal imbalance with drastic consequences for oocyte maturation and embryo development. Firstly, maternal obesity was shown to alter insulin, glucose, and free fatty acid concentration in follicular fluid, directly affecting oocyte metabolism and reducing oocyte maturation [29]. Secondly, insulin-stimulated glucose uptake was shown to be impaired in CC isolated from mice treated with HFD, suggesting the establishment of insulin-resistance [30]. Indeed, activation of the polyol pathway during hyperglycemia was shown to negatively affect metabolism and CC–oocyte communication [31]. As a result, in obese mothers, oocyte maturation, fertilization rate, and embryo quality were significantly decreased [32]. Moreover, oocytes derived from obese mice after in vitro fertilization and culture presented reduced development [32]. Furthermore, studies in mice clearly linked maternal obesity with oocyte and zygote increased mitochondrial potential, mitochondrial DNA content and biogenesis, and generation of reactive oxygen species (ROS) [33]. Importantly, the absence of mitophagy was presented as the main cause of mitochondrial dysfunction in oocytes and early embryos from obese mothers [34]. Another recent report also linked lack of expression of Stella in oocytes from obese mice with increased hydromethylation in the zygote and DNA instability [35]. Thus, obesity can drastically affect both oocyte and embryo quality.
4. Conclusion
Folliculogenesis regulation is a complex process that depends on the crosstalk between local and systemic factors, accounting, therefore, for its vulnerability to maternal physiological fitness. Indeed, leptin, an established local regulator of folliculogenesis, presents increased circulating levels in obese mothers with major consequences for follicular activation, recruitment, and growth. In addition to increased ovarian ObRb activation and altered leptin signaling, deleterious effects of systemic hyperleptinemia during obesity can also result from local overexpression of mediators of leptin resistance, such as SOCS3. As a result, leptin’s prominent role as suppressor of follicular pool activation can be affected in obese mothers, with consequent POF. During preovulatory follicle formation, altered leptin signaling affects not only steroidogenesis but the communication between GC and oocyte, which is known to be key for antrum formation and oocyte growth. Finally, changes in leptin signaling and impaired metabolism may also incur drastic consequences for oocyte maturation, hampering meiosis resumption and cytoplasmic maturation (Figure 3). Hence, advances in our understanding of the role of leptin on ovarian pathophysiology during obesity should unravel innovative tools to monitor the quality of the oocyte during disease progression, potentially preventing pregnancy failure and ensuring the birth of a healthy offspring.
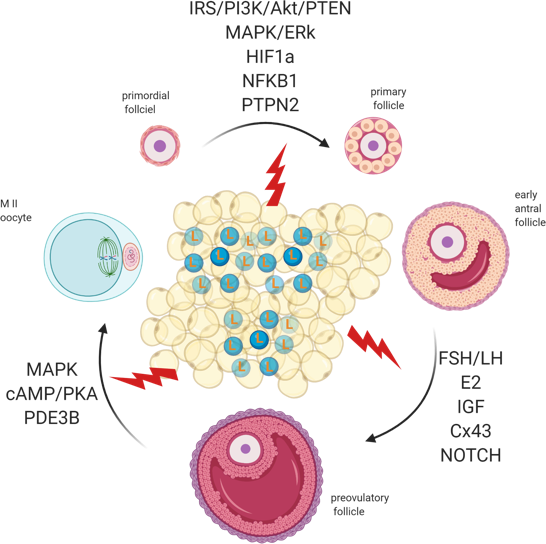
Figure 3. Adipose tissue secretes excessive amounts of leptin (L) during obesity. Leptin signaling in the ovaries of obese mothers is altered, culminating with the establishment of leptin resistance. As a result, signaling pathways governing primordial follicle activation, preovulatory follicle formation, and oocyte maturation can be affected. Created with BioRender.com, accessed on 1 November 2020.
References
- Gambineri, A.; Laudisio, D.; Marocco, C.; Radellini, S.; Colao, A.; Savastano, S. Female infertility: Which role for obesity? Int. J. Obes. Suppl. 2019, 9, 65–72.
- Dag, Z.O.; Dilbaz, B. Impact of obesity on infertility in women. J. Turk. Gynecol. Assoc. 2015, 16, 111–117.
- Cree-Green, M.; Newcomer, B.R.; Coe, G.; Newnes, L.; Baumgartner, A.; Brown, M.S.; Pyle, L.; Reusch, J.E.; Nadeau, K.J. Peripheral insulin resistance in obese girls with hyperandrogenism is related to oxidative phosphor-ylation and elevated serum free fatty acids. Am. J. Physiol. Metab. 2015, 308, E726–E733.
- Chang, A.S.; Dale, A.N.; Moley, K.H. Maternal Diabetes Adversely Affects Preovulatory Oocyte Maturation, Development, and Granulosa Cell Apoptosis. Endocrinology 2005, 146, 2445–2453.
- Skaznik-Wikiel, M.E.; Swindle, D.C.; Allshouse, A.A.; Polotsky, A.J.; McManaman, J.L. High-Fat Diet Causes Subfertility and Compromised Ovarian Function Independent of Obesity in Mice1. Biol. Reprod. 2016, 94, 108.
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild–type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97.
- Odle, A.K.; Akhter, N.; Syed, M.M.; Allensworth-James, M.L.; Beneš, H.; Castillo, A.I.M.; MacNicol, M.C.; MacNicol, A.M.; Childs, G.V. Leptin Regulation of Gonadotrope Gonadotropin-Releasing Hormone Receptors as a Metabolic Checkpoint and Gateway to Reproductive Competence. Front. Endocrinol. 2018, 8, 367.
- Ryan, N.K.; Woodhouse, C.M.; Van Der Hoek, K.H.; Gilchrist, R.B.; Armstrong, D.T.; Norman, R.J. Expression of leptin and its receptor in the murine ovary: Possible role in the regulation of oocyte maturation. Biol. Reprod. 2002, 66, 1548–1554.
- Karlsson, C.; Lindell, K.; Svensson, E.; Bergh, C.; Lind, P.; Billig, H.; Carlsson, L.M.; Carlsson, B. Expression of Functional Leptin Receptors in the Human Ovary 1. J. Clin. Endocrinol. Metab. 1997, 82, 4144–4148.
- Welt, C.K.; Schneyer, A.L.; Heist, K.; Mantzoros, C.S. Leptin and Soluble Leptin Receptor in Follicular Fluid. J. Assist. Reprod. Genet. 2003, 20, 495–501.
- Maffei, M.; Halaas, J.L.; Ravussin, E.; Pratley, R.E.; Lee, G.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161.
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.-M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nat. Cell Biol. 1998, 392, 398–401.
- Tu, X.; Kuang, Z.; Gong, X.; Shi, Y.; Yu, L.; Shi, H.; Wang, J.; Sun, Z. The Influence of LepR Tyrosine Site Mutations on Mouse Ovary Development and Related Gene Expression Changes. PLoS ONE 2015, 10, e0141800.
- Bilbao, M.G.; Di Yorio, M.P.; Faletti, A.G. Different levels of leptin regulate different target enzymes involved in proges-terone synthesis. Fertil. Steril. 2013, 99, 1460–1466.
- Galvão, A.; Tramontano, A.; Rebordão, M.R.; Amaral, A.; Bravo, P.P.; Szóstek, A.; Skarzynski, D.; Mollo, A.; Ferreira-Dias, G. Opposing Roles of Leptin and Ghrelin in the Equine Corpus Luteum Regulation: An In Vitro Study. Mediat. Inflamm. 2014, 2014, 1–13.
- Panwar, S.; Herrid, M.; Kauter, K.G.; McFarlane, J.R. Effect of passive immunization against leptin on ovarian follicular development in prepubertal mice. J. Reprod. Immunol. 2012, 96, 19–24.
- Swain, J.E.; Dunn, R.L.; McConnell, D.; Gonzalez-Martinez, J.; Smith, G.D. Direct Effects of Leptin on Mouse Reproductive Function: Regulation of Follicular, Oocyte, and Embryo Development. Biol. Reprod. 2004, 71, 1446–1452.
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Chang, H.-M.; Yu, Y.; Wang, T.; Lu, C.; Yang, M.; Yang, S.; et al. Transcriptome Landscape of Human Folliculogenesis Reveals Oocyte and Granulosa Cell Interactions. Mol. Cell 2018, 72, 1021–1034.e4.
- Wołodko, K.; Walewska, E.; Adamowski, M.; Castillo-Fernandez, J.; Kelsey, G.; Galvão, A. Leptin Resistance in the Ovary of Obese Mice is Associated with Profound Changes in the Transcriptome of Cumulus Cells. Cell. Physiol. Biochem. 2020, 54, 417–437.
- He, Y.; Tian, J.; Blizzard, L.; Oddy, W.H.; Dwyer, T.; Bazzano, L.A.; Hickey, M.; Harville, E.W.; Venn, A.J. Associations of childhood adiposity with menstrual irregularity and polycystic ovary syndrome in adulthood: The Childhood Determinants of Adult Health Study and the Bogalusa Heart Study. Hum. Reprod. 2020, 35, 1185–1198.
- Wang, N.; Luo, L.-L.; Xu, J.-J.; Xu, M.-Y.; Zhang, X.-M.; Zhou, X.-L.; Liu, W.-J.; Fu, Y.-C. Obesity accelerates ovarian follicle development and follicle loss in rats. Metabolism 2014, 63, 94–103.
- Nteeba, J.; Ross, J.; Ii, J.P.; Keating, A. High fat diet induced obesity alters ovarian phosphatidylinositol-3 kinase signaling gene expression. Reprod. Toxicol. 2013, 42, 68–77.
- Hegyi, K.; Fülöp, K.; Kovacs, K.; Tóth, S.; Falus, A. Leptin-induced signal transduction pathways. Cell Biol. Int. 2004, 28, 159–169.
- Xia, H.; Zhang, R.; Guan, H.; Zhang, W. Follicle loss and PTEN/PI3K/mTOR signaling pathway activated in LepR-mutated mice. Gynecol. Endocrinol. 2019, 35, 44–48.
- Quinkler, M.; Sinha, B.; Tomlinson, J.W.; Bujalska, I.J.; Stewart, P.M.; Arlt, W. Androgen generation in adipose tissue in women with simple obesity—A site-specific role for 17β-hydroxysteroid dehydrogenase type 5. J. Endocrinol. 2004, 183, 331–342.
- Wu, S.; Divall, S.; Nwaopara, A.; Radovick, S.; Wondisford, F.; Ko, C.; Wolfe, A. Obesity-Induced Infertility and Hyperandrogenism Are Corrected by Deletion of the Insulin Receptor in the Ovarian Theca Cell. Diabetes 2013, 63, 1270–1282.
- Souter, I.; Baltagi, L.M.; Kuleta, D.; Meeker, J.D.; Petrozza, J.C. Women, weight, and fertility: The effect of body mass index on the outcome of superovulation/intrauterine insemination cycles. Fertil. Steril. 2011, 95, 1042–1047.
- Sagae, S.C.; Menezes, E.F.; Bonfleur, M.L.; Vanzela, E.C.; Zacharias, P.; Lubaczeuski, C.; Franci, C.R.; Sanvitto, G.L. Early onset of obesity induces reproductive deficits in female rats. Physiol. Behav. 2012, 105, 1104–1111.
- Purcell, S.H.; Moley, K.H. The impact of obesity on egg quality. J. Assist. Reprod. Genet. 2011, 28, 517–524.
- Purcell, S.H.; Chi, M.M.; Moley, K.H. Insulin-Stimulated Glucose Uptake Occurs in Specialized Cells within the Cumulus Oocyte Complex. Endocrinology 2012, 153, 2444–2454.
- Sutton-McDowall, M.L.; Gilchrist, R.B.; Thompson, J.G. The pivotal role of glucose metabolism in determining oocyte developmental competence. Reproduction 2010, 139, 685–695.
- Robker, R.L. Evidence that obesity alters the quality of oocytes and embryos. Pathophysiology 2008, 15, 115–121.
- Igosheva, N.; Abramov, A.Y.; Poston, L.; Eckert, J.J.; Fleming, T.P.; Duchen, M.R.; McConnell, J. Maternal Diet-Induced Obesity Alters Mitochondrial Activity and Redox Status in Mouse Oocytes and Zygotes. PLoS ONE 2010, 5, e10074.
- Boudoures, A.L.; Saben, J.; Drury, A.; Scheaffer, S.; Modi, Z.; Zhang, W.; Moley, K.H. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev. Biol. 2017, 426, 126–138.
- Han, L.; Ren, C.; Li, L.; Li, X.; Ge, J.; Wang, H.; Miao, Y.-L.; Guo, X.; Moley, K.H.; Shu, W.; et al. Embryonic defects induced by maternal obesity in mice derive from Stella insufficiency in oocytes. Nat. Genet. 2018, 50, 432–442.

