 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maolin Lu | + 1540 word(s) | 1540 | 2021-04-25 05:54:00 | | | |
| 2 | Catherine Yang | Meta information modification | 1540 | 2021-05-11 03:25:33 | | |
Video Upload Options
Single-molecule Förster resonance energy transfer (smFRET) has provided a powerful platform to connect structure–function in motion, revealing dynamic aspects of spikes for several viruses: SARS-CoV-2, HIV-1, influenza, and Ebola.
1. Introduction
Virus spikes on the surface of enveloped viruses are often also viral fusion proteins that mediate the fusion between viral membranes and cellular membranes (Figure 1) essential for virus entry [1][2][3]. The merging of virus and lipid bilayers progresses through a hemifusion intermediate, followed by a fusion pore widening, content mixing, and the delivery of virus capsids into the host cytoplasm [4]. Viral fusion proteins respond to the binding of cellular receptors or acidic pH to undergo conformational rearrangements, which eventually promote membrane fusion. Viral fusion proteins have been categorized into three classes [1][2], of which Class I viral fusion proteins include the medically important SARS-CoV-2 spike (S) protein, the HIV-1 envelope (Env) protein, influenza hemagglutinin (HA), and Ebola glycoprotein (GP). These virus spikes are first synthesized as trimers of a single-chain polypeptide—an immature precursor, then go through proteolytical processing by host proteases to form mature spikes—trimers of heterodimers (Figure 1A). Mature spikes are highly metastable on the virus surface. Upon interacting with hosts, mature spikes undergo conformational changes from pre-fusion conformations to the lowest-energy post-fusion conformation (a common hairpin-like or the analogous coiled-coil conformation) through hypothetical intermediates in which the fusion peptide extends and inserts into the host target membrane (Figure 1B). Numerous pre-fusion and post-fusion structures of virus spikes have provided unprecedented details of conformations at individual steps during the viral membrane fusion [5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21]. The recent dynamic studies on virus spikes established platforms to connect these structural snapshots in real time, revealed the order and the kinetics of transitional events, and guided developing interventions aiming to arrest or block viral membrane fusion, thus stopping viral infection [22][23][24][25][26][27][28][29][30][31].
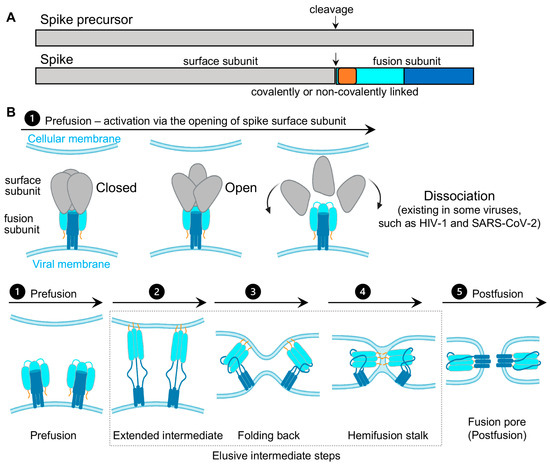
As spikes are highly exposed to our immune system, they are main targets of neutralizing antibodies and thus are critical for developing vaccines and anti-spike therapeutics. Most vaccines or vaccine candidates for HIV-1/AIDS and SARS-CoV-2/COVID-19 are based on their spike proteins to trigger the immune system to produce neutralizing antibodies. Interestingly, in the face of immune pressure, many viral spike proteins use conformational masking of vulnerable antibody-targeted epitopes. In addition to glycan shields and hypermutations, this strategy of conformational masking has been best-understood in HIV-1 [32][33][34][35][36][37].
Collectively, virus spike proteins are structurally flexible and conformationally dynamic on the virus surface. The dynamic nature and conformational plasticity of spikes enables entry of enveloped viruses into host target cells through membrane fusion, while distracting the immune system from antibody recognition. Molecular understanding and modulation of spikes’ intrinsic dynamics can guide the rational design of spike-targeting interventions such as vaccines, small-molecule inhibitors, and antibodies to block virus entry. Single-molecule Förster resonance energy transfer (smFRET), a spectroscopic tool sensitive to inter-fluorophore molecular distances, has been well-positioned to capture the innate dynamics of virus spikes labeled with Förster resonance energy transfer (FRET)-paired fluorophores. This minireview covers recent work on single-molecule FRET imaging of class I fusion proteins of enveloped viruses, including SARS-CoV-2 [31], HIV-1 [22][23][24][25][26][27], influenza [29], and Ebola [28][30].
2. Single-Molecule Förster Resonance Energy Transfer (smFRET) Imaging
Imaging macromolecules at the single-molecule/single-particle level has advanced our understanding of both static and dynamic aspects in virus–host interactions, merited by avoiding the averaging-out effect from traditional ensemble-level measurements. Those imaging techniques, such as single-particle cryoEM/cryoET, single-particle optical tracking, and super-resolution fluorescence microscopy exerted significant roles in addressing fundamental questions with regards to structures, dynamics, and functions of virus molecules underlying virus–host interactions [38][39][40][41][42][43][44].
Single-molecule Förster resonance energy transfer (smFRET) imaging has been proven to be a reliable imaging tool for revealing the intrinsically dynamic, heterogeneous nature of biological systems [45][46][47]. It is powerful to probe protein conformational dynamics in a time-resolved manner; identify previously unknown states; detect intermediates transient in nature; and delineate the sequence, the order, the timing, the frequency of conformational states, and the transitional events. Two significant advantages of smFRET are (1) it permits direct in situ observations of different conformations/structures of biological molecules in real time, and (2) it reveals conformational intermediates and features that are previously concealed or averaged-out in ensembles.
FRET refers to non-radiative energy transfer between a donor and acceptor fluorophore. The energy transfer efficiency is a function of distances between both fluorophores, described as FRET = 1/(1 + (R/R0)6), where R is the inter-fluorophore distance and R0 is the Förster distance determining the range of sensitive measure of distance [47]. For mostly used paired-fluorophores, the FRET-detectable distance ranges from 30 Å to 80 Å, well suited for the dimensions of spike proteins of enveloped viruses (Figure 2A). In application, a pair of donor and acceptor fluorophores is site-specifically labeled on the one or two molecules of interest. The donor fluorophores are excited by a laser, and the single-molecule fluorescence from both fluorophores is separately recorded for seconds to minutes by total internal fluorescence microscope (TIRF) or confocal microscopy [47], in which prism-based TIRF and objective TIRF are widely used (Figure 2B). Fluorescence and the quantified FRET values monitor the proximity between two fluorophores in real-time, ultimately translating to the object’s intra-molecular or inter-molecular dynamics (Figure 2C,D). The object of interest can be genetic materials, proteins, peptides, and other biomolecules. The applications of FRET in biological systems are broad, thanks to advances in instrumentations, analysis software, and site-specific dye-labeling methods. For instance, newly developed scientific CMOS (sCMOS) cameras and the developed smFRET software or algorisms [48][49][50][51] facilitate high-throughput smFRET imaging and robust data analysis.
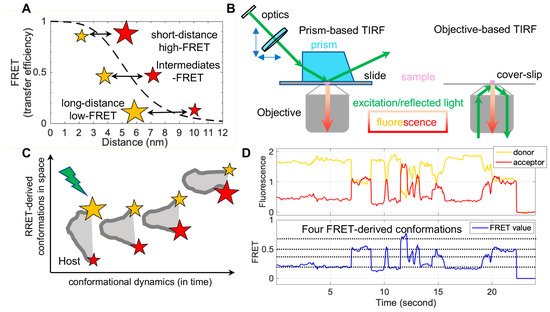
In contrast, the attachment of fluorophores to specific sites on proteins without disturbing their functionality has been a technical obstacle. The conventional way to label proteins is to introduce cysteines, which permit maleimide-functionalized dyes’ attachments [52]. A considerable number of pre-existing essential cysteines on virus proteins, especially spike proteins, make it infeasible. Two alternative fluorophore-attaching strategies, enzymatic and amber-click labeling, have overcome this hurdle. Enzymatic methods take advantage of enzymes that site-specifically recognize short-peptide tags (6–12 amino acids in length) introduced into the protein of interest and transfer dye-conjugated substrates to these tags [53][54]. The genetically encoded copper-free click chemistry (amber-click) allows reading through introduced amber stop codons on the protein of interest as unnatural amino acids through amber suppression, followed by site-specifically labeling conjugated dyes on unnatural amino acids by copper-free click chemistry [55][56]. Both enzymatic and amber-click methods have been applied to study virus spikes for many enveloped viruses to reveal dynamic aspects during viral membrane fusion [22][23][24][25][26][27][28][29][30][31].
References
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698.
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507.
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122.
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683.
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592.
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263.
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273.
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430.
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659.
- Julien, J.P.; Cupo, A.; Sok, D.; Stanfield, R.L.; Lyumkis, D.; Deller, M.C.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1477–1483.
- Lyumkis, D.; Julien, J.P.; de Val, N.; Cupo, A.; Potter, C.S.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; Carragher, B.; et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1484–1490.
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373.
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832.
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43.
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 2006, 312, 404–410.
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929.
- Weissenhorn, W.; Carfi, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616.
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc. Natl. Acad. Sci. USA 1999, 96, 2662–2667.
- Falzarano, D.; Krokhin, O.; Wahl-Jensen, V.; Seebach, J.; Wolf, K.; Schnittler, H.J.; Feldmann, H. Structure-function analysis of the soluble glycoprotein, sGP, of Ebola virus. Chembiochem 2006, 7, 1605–1611.
- Misasi, J.; Gilman, M.S.; Kanekiyo, M.; Gui, M.; Cagigi, A.; Mulangu, S.; Corti, D.; Ledgerwood, J.E.; Lanzavecchia, A.; Cunningham, J.; et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 2016, 351, 1343–1346.
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763.
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 2018, 7, e34271.
- Alsahafi, N.; Bakouche, N.; Kazemi, M.; Richard, J.; Ding, S.; Bhattacharyya, S.; Das, D.; Anand, S.P.; Mothes, W.; Lifson, J.; et al. An asymmetric opening of HIV-1 Env is required for anti-cluster A antibody binding. Cell Host Microbe 2019, 25, 578–587.
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419.
- Li, Z.; Li, W.; Lu, M.; Bess, J., Jr.; Chao, C.W.; Gorman, J.; Terry, D.S.; Zhang, B.; Zhou, T.; Blanchard, S.C.; et al. Subnanometer structures of HIV-1 envelope trimers on aldrithiol-2-inactivated virus particles. Nat. Struct. Mol. Biol. 2020, 27, 726–734.
- Lu, M.; Ma, X.; Reichard, N.; Terry, D.S.; Arthos, J.; Smith, A.B., 3rd; Sodroski, J.G.; Blanchard, S.C.; Mothes, W. Shedding-Resistant HIV-1 Envelope Glycoproteins Adopt Downstream Conformations That Remain Responsive to Conformation-Preferring Ligands. J. Virol. 2020, 94.
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020, 18, e3000626.
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018, 174, 926–937.e912.
- Durham, N.D.; Howard, A.R.; Govindan, R.; Senjobe, F.; Fels, J.M.; Diehl, W.E.; Luban, J.; Chandran, K.; Munro, J.B. Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics. Viruses 2020, 12, 103.
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880–891.e888.
- Wyatt, R.; Kwong, P.D.; Desjardins, E.; Sweet, R.W.; Robinson, J.; Hendrickson, W.A.; Sodroski, J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393, 705–711.
- Kwong, P.D.; Doyle, M.L.; Casper, D.J.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682.
- Crispin, M.; Ward, A.B.; Wilson, I.A. Structure and immune recognition of the HIV glycan shield. Annu. Rev. Biophys. 2018, 47, 499–523.
- Lewis, G.K.; Finzi, A.; DeVico, A.L.; Pazgier, M. Conformational Masking and Receptor-Dependent Unmasking of Highly Conserved Env Epitopes Recognized by Non-Neutralizing Antibodies That Mediate Potent ADCC against HIV-1. Viruses 2015, 7, 5115–5132.
- Kwong, P.D.; Mascola, J.R.; Nabel, G.J. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: The end of the beginning. Nat. Rev. Immunol. 2013, 13, 693–701.
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312.
- Stass, R.; Ng, W.M.; Kim, Y.C.; Huiskonen, J.T. Structures of enveloped virions determined by cryogenic electron microscopy and tomography. Adv. Virus Res. 2019, 105, 35–71.
- Goetschius, D.J.; Lee, H.; Hafenstein, S. CryoEM reconstruction approaches to resolve asymmetric features. Adv. Virus Res. 2019, 105, 73–91.
- Lu, M.; Ma, X.; Mothes, W. Illuminating the virus life cycle with single-molecule FRET imaging. Adv. Virus Res. 2019, 105, 239–273.
- Howard, A.R.; Munro, J.B. Developments in single-molecule and single-particle fluorescence-based approaches for studying viral envelope glycoprotein dynamics and membrane fusion. Adv. Virus Res. 2019, 104, 123–146.
- Liu, S.L.; Wang, Z.G.; Xie, H.Y.; Liu, A.A.; Lamb, D.C.; Pang, D.W. Single-Virus Tracking: From Imaging Methodologies to Virological Applications. Chem. Rev. 2020, 120, 1936–1979.
- Groves, N.S.; Bruns, M.M.; van Engelenburg, S.B. A Quantitative Live-Cell Superresolution Imaging Framework for Measuring the Mobility of Single Molecules at Sites of Virus Assembly. Pathogens 2020, 9, 972.
- Iliopoulou, M.; Nolan, R.; Alvarez, L.; Watanabe, Y.; Coomer, C.A.; Jakobsdottir, G.M.; Bowden, T.A.; Padilla-Parra, S. A dynamic three-step mechanism drives the HIV-1 pre-fusion reaction. Nat. Struct. Mol. Biol. 2018, 25, 814–822.
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972.
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359, eaan1133.
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516.
- McKinney, S.A.; Joo, C.; Ha, T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006, 91, 1941–1951.
- Juette, M.F.; Terry, D.S.; Wasserman, M.R.; Altman, R.B.; Zhou, Z.; Zhao, H.; Blanchard, S.C. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat. Methods 2016, 13, 341–344.
- Thomsen, J.; Sletfjerding, M.B.; Jensen, S.B.; Stella, S.; Paul, B.; Malle, M.G.; Montoya, G.; Petersen, T.C.; Hatzakis, N.S. DeepFRET, a software for rapid and automated single-molecule FRET data classification using deep learning. Elife 2020, 9.
- van de Meent, J.W.; Bronson, J.E.; Wiggins, C.H.; Gonzalez, R.L., Jr. Empirical Bayes methods enable advanced population-level analyses of single-molecule FRET experiments. Biophys. J. 2014, 106, 1327–1337.
- Kim, Y.; Ho, S.O.; Gassman, N.R.; Korlann, Y.; Landorf, E.V.; Collart, F.R.; Weiss, S. Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 2008, 19, 786–791.
- Lin, C.W.; Ting, A.Y. Transglutaminase-catalyzed site-specific conjugation of small-molecule probes to proteins in vitro and on the surface of living cells. J. Am. Chem. Soc. 2006, 128, 4542–4543.
- Yin, J.; Lin, A.J.; Golan, D.E.; Walsh, C.T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 2006, 1, 280–285.
- Plass, T.; Milles, S.; Koehler, C.; Schultz, C.; Lemke, E.A. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. Engl. 2011, 50, 3878–3881.
- Sakin, V.; Hanne, J.; Dunder, J.; Anders-Osswein, M.; Laketa, V.; Nikic, I.; Krausslich, H.G.; Lemke, E.A.; Muller, B. A versatile tool for live-cell imaging and super-resolution nanoscopy studies of HIV-1 Env distribution and mobility. Cell Chem. Biol. 2017, 24, 635–645.

