 +1 credit
+1 credit
Video Upload Options
Acetylcholinesterase (AChE) inhibitors are the only drugs that have demonstrated success in slowing shrinkage (atrophy) of the cortex, hippocampus, and basal forebrain, major areas of Alzheimer's disease (AD)-associated brain damage and dementia. The main barrier to taking advantage of this new success in treating, or even preventing, AD is that the old available AChE inhibitors are weak reversible inhibitors that cause intolerable nausea, vomiting, and diarrhea if given in the higher doses needed. A promising new strategy for producing high-level AChE inhibition in the brain as needed for effective treatment of AD is the use of AChE inhibitors that are of a different type, the irreversible inhibitors.
1. AChE Inhibitors and Anticholinergics Affect Neurodegeneration in AD
Degeneration of basal forebrain neurons causes a loss of cholinergic tone in the basal forebrain cholinergic system, especially projections to the cortex and hippocampus, which is responsible for the severe cognitive losses characteristic of AD [1][2][3][4][5]. The magnocellular neurons of the basal forebrain are among the earliest to undergo severe neurodegeneration in AD [6]. Atrophy of these neurons occurs during normal aging and early in the progression of AD [5][7]. In vivo longitudinal imaging studies indicate that degeneration of the basal forebrain in prodromal AD precedes and predicts entorhinal pathology and memory impairment [8]. Changes in basal forebrain volume is also a reliable indicator of cortical spread of AD-induced neurodegeneration, which supports the contention that basal forebrain neurodegeneration is an upstream triggering event in the development of AD [9]. Atrophy of the basal forebrain, in particular, also predicts cortical amyloid burden [10]. Degeneration of the basal forebrain in preclinical, but cognitively normal suspected prodromal AD, is associated with increased microglial inflammation and amyloid and tau accumulation in vivo at the earliest stages of the disease, which suggests that the loss of central cholinergic tone from the basal forebrain may enable microglial inflammation induced by amyloid and tau accumulation [11]. The cholinergic neurons of the basal forebrain are also among the earliest to show tauopathy, the oligomeric constituents of neurofibrillary tangles in AD [12][13][14][15]. Atrophy of the basal forebrain, in particular, predicts the development of AD in the asymptomatic elderly [16]. Evidence now suggests that the cholinergic cell bodies of the basal forebrain are not completely lost in AD, but that many persist in an atrophied state in which they have lost their cholinergic phenotype [2]. Thus, the collapse of basal forebrain neurons, including loss of their projection fibers and the subsequent absence of their synaptic acetylcholine efflux and cholinergic tone in the cortex and hippocampus, may be a germinal event in the development of AD [2][3][4][5][7][10][12].
The key role of cholinergic tone is confirmed by animal experiments in which basal forebrain lesions (an animal model of AD) or by treatment with anticholinergics (blocking acetylcholine receptors) triggers the formation of β-amyloid in transgenic mice [17][18], rats [19], guinea pigs [20], and rabbits [21]. These animal models suggest that all or most normal (non-transgenic) mammalian brains have an incipient age-related capacity to produce amyloid like that which occurs spontaneously in aged primates [22]. Furthermore, the extent of amyloid production occurs on a continuum that is substantially skewed upward toward older animals, those with basal forebrain cholinergic lesions, and those with transgenic with human amyloid-related genes [17][18][19][20][21][22]. The importance of AChE inhibitors, which restore cholinergic function by amplifying the effect of synaptic acetylcholine, is shown by the fact that they are prophylactic against some of these changes [21][23].
In humans, the long term use of anticholinergics triggers the accumulation of both plaques and tangles as seen on postmortem examination [24]. Anticholinergics also accelerate the progression from normal cognition to more advanced stages of mild cognitive impairment and conversion into AD-like dementia in elderly persons [25][26][27][28]. In addition, AChE inhibition, an independent effect separate from memantine, slows the clinical progression of AD [29][30][31][32][33][34][35].
The anti-neurodegenerative benefits of AChE inhibition on CNS atrophy, a direct biomarker of AD pathophysiology, are more convincing. For example, in a retrospective analysis patients with mild cognitive impairment, rivastigmine, which inhibits both AChE and butyrylcholinesterase (BChE, EC 3.1.1.8) [36], reduces whole brain atrophy, hippocampal atrophy, and white matter loss [37]. In another study, 20 weeks of treatment with rivastigmine protected against AD-associated white matter loss, an effect that was not observed with donepezil and galantamine, more AChE-selective inhibitors [36]. Rivastigmine-associated protection of white matter is attributed to BChE inhibition [38] and the role of cholinergic signaling, especially involving BChE and its presence in white matter [39][40][41], but rivastigmine is also a potent inhibitor of AChE [36] and such an attribution deserves further study. More specific to AChE, however, randomized, placebo-controlled trials show that short-term donepezil-induced AChE inhibition (one year) in prodromal AD patients slows gray matter atrophy in the hippocampus [42], cortex [43], and basal forebrain [44]. Donepezil-induced AChE inhibition (six months) in patients who have advanced to mild or moderate AD also slows hippocampal atrophy [45]. The anti-neurodegenerative benefits of AChE inhibition on the basal forebrain and its projection areas (hippocampus and cortex) in AD are clear.
The mechanism(s) by which AChE inhibitors produce these disease-modifying benefits are not clear. One hypothesis is that the AChE inhibitors act by enhancing neurotrophic factors, especially nerve growth factor (NGF), which affect key AD-associated pathophysiological processes in the basal forebrain, cortex, and hippocampus [2][46][47][48][49]. The effects of NGF and its possible role in AD, the neurotrophic hypothesis of AD [50][51], and the extensive supporting evidence, have been reviewed in detail elsewhere [2][47]. Briefly, the AD-associated loss of basal forebrain cholinergic neurons, or their cholinergic phenotype, results in a loss of acetylcholine-dependent stimulation of the production and release of NGF from the basal forebrain target tissues (hippocampus and cortex). With declining acetylcholine stimulation, there is a resulting deficit of mature NGF for uptake into the presynaptic terminals of the cholinergic projection axons and inadequate NGF undergoing microtubule retrograde transport back to the basal forebrain cholinergic cell bodies. Without adequate NGF trophic effects, the basal forebrain cholinergic neurons atrophy or lose their cholinergic phenotype [2]. In this scenario, AChE inhibitors amplify acetylcholine-dependent stimulation and release of NGF and, thereby, increase the survival of the basal forebrain cholinergic system, an anti-neurodegenerative effect [2][47]. The role of the basal forebrain cholinergic system, neurotrophic factors, and alternative hypotheses such as tauopathy and inflammation are not mutually exclusive but contribute converging insights into the pathogenesis of AD [48]. Regardless of the mechanism of AChE inhibitor-induced anti-neurodegenerative benefit, there is a call for more effective CNS cholinergic stimulation to improve disease-modifying benefits in AD therapy [2][52].
In summary, increasing cholinergic tone (AChE inhibition) or deceasing cholinergic tone (anticholinergics) produce disease-modifying effects by either slowing or accelerating, respectively, the clinical and pathophysiological progression of AD. In view of the decades of failures of other disease-modifying strategies and the critical need for effective treatments, AChE inhibitors offer an unparalleled opportunity for delaying the onset, slowing the disease, reducing disability and preserving the autonomy of patients at risk for AD.
2. Failures of Current AChE Inhibitors
The rationale for the use of AChE inhibitors is to stop the breakdown of synaptic acetylcholine, amplify and extend its impact in the basal forebrain cholinergic system, and to enhance the cholinergic and cognitive functions which deteriorate in normal aging and AD [1][2][3][4][5][53][54][55][56]. While the use of AChE inhibitors has a clear rational basis, their impact on cognitive functions, quality of life, global clinical states, and medicoeconomic benefits are marginal to nonexistent and have fallen short of expectations [57][58][59][60][61]. Even though the currently available AChE inhibitors (donepezil, rivastigmine, galantamine) have produced the most robust anti-neurodegenerative benefits to date [37][38][39][40][41][42][43][44][45], their effects are small and are of more theoretical interest than clinical importance [62][63]. The current AChE inhibitors are far from adequate to meet the demand for highly effective AD interventions [64] that are urgently needed to improve cognitive functions and/or take advantage of the recently recognized additional anti-neurodegenerative benefits [37][38][39][40][41][42][43][44][45].
The main limitation of the current AChE inhibitors is the unavoidable gastrointestinal toxicity that limits their use to doses that are too low to be effective [52]. Direct PET measurements of the maximum in vivo cortical AChE inhibition that can be tolerated in AD patients undergoing donepezil treatment is estimated at ~19% [65], ~27% [66], ~35% [67], and from 28% to 39%, depending on the cortical area [68]. Similarly, in vivo cortical AChE inhibition during rivastigmine and galantamine treatment is estimated at ~28% to 37% [68] and 30% to 40% [69], respectively. This level of AChE inhibition, as found in clinical use, is less than the minimum of ~50% AChE inhibition required for effective AD therapy [52][70][71][72]. In view of these data, it is not surprising that AChE inhibitors produce mainly statistical improvements in cognitive function, but certainly not the powerful clinical improvements that were originally expected [62][63][64]. On the other hand, AChE-induced anti-neurodegenerative benefits are unexpected under such severely limiting circumstances as low levels of inhibition in (25–35%), short-term trials (6 months to one year), and with only a few hundred patients in each experiment [42][43][44][45].
It is reasonable to speculate that a broad range of improvement in AChE therapy, high-level AChE inhibition above 50%, could substantially improve anti-neurodegenerative outcomes, but only if the long-time barrier to dose-limiting gastrointestinal toxicity can be overcome [52].
3. Mechanisms of Action of Key AChE Inhibitors
The mechanisms by which AChE inhibitors block the catalytic action of the enzyme fall into three major categories: competitive inhibition, pseudo-irreversible inhibitions, and irreversible inhibition. The most important AChE inhibitors used for the treatment of AD are shown in Table 1.
Table 1. AChE Inhibitors.
| Inhibitor | Mechanism of Action | Additional Notes * |
|---|---|---|
| Donepezil | Competitive/Noncompetitive | |
| Galantamine | Competitive | Upregulates nicotinic receptors |
| Rivastigmine | Pseudo-Irreversible | Also inhibits BChE |
| Metrifonate | Pseudo-Irreversible | Induces peripheral neuropathy |
| Methanesulfonyl Fluoride | Irreversible | High CNS Selectivity |
A schematic representation of how acetylcholine and the key AChE inhibitors interact with the catalytic action of the enzyme is shown in Figure 1:
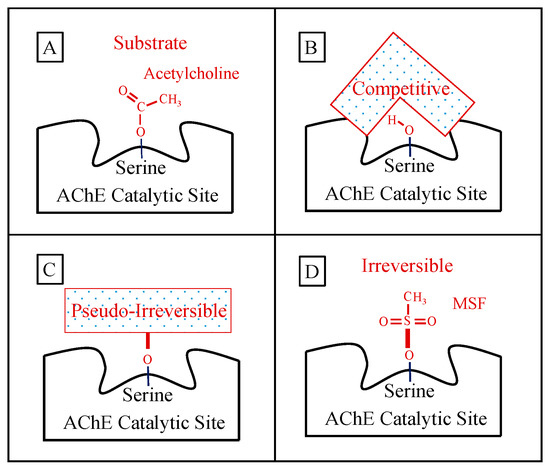
Figure 1. Panel (A) shows the normal ephemeral (microseconds) covalent acetyl-enzyme complex that is formed as an intermediate step in the hydrolysis of acetylcholine (shown). Panel (B) shows a schematic of a competitive inhibitor binding reversibly (spanning) the catalytic site representing donepezil or galantamine (note that the competitive inhibitor does NOT form a covalent bond with the serine sidechain OH required for acetylcholine hydrolysis). Panel (C) shows a longer-lasting covalent bond (signified by heavier red bars) formed between pseudo-irreversible inhibitors (spontaneously hydrolyzed with a half-time of hours) and the enzyme. The schematic box in Panel (C) represents the corresponding carbamoyl- or phosphoryl-enzyme covalent binding, respectively, for the case of rivastigmine, metrifonate, or DDVP, wherein the specific molecular structure of each pseudo-irreversible inhibitor intermediate not shown. Panel (D) shows an example of the irreversible sulfonyl-enzyme covalent complex (no spontaneous hydrolysis, no recovery) that permanently excludes acetylcholine binding and hydrolysis. The specific sulfonyl-enzyme covalent complex shown in Panel (D) is that formed during methanesulfonyl fluoride inhibition.
3.1. Mechanisms of Action of Short-Acting AChE Inhibitors—Limited Efficacy
Details of the mechanisms of action of the current AChE inhibitors (donepezil, rivastigmine, and galantamine) have been reviewed in detail elsewhere [62][72]. Briefly, as shown in Table 1, the mechanisms of action of the available short-acting reversible AChE inhibitors fall into two categories: competitive or pseudo-irreversible inhibition.
3.1.1. Competitive AChE Inhibition: Donepezil and Galantamine
Competitive inhibition is dependent upon the concentration of the inhibitor in the microenvironment of the enzyme in the synapse and the degree to which the inhibitor occupies the catalytic site. It is readily reversible with declining in vivo inhibitor concentration, and, therefore, the duration of the inhibition by donepezil and galantamine is dependent on the rate of inhibitor elimination [62][72].
Donepezil (FDA 1996, Aricept®) is a mixed competitive/noncompetitive inhibitor that binds to and orients over the catalytic gorge as well as spans a peripheral binding site which both directly and indirectly blocks catalytic action [73][74]. Donepezil disappears from blood with an elimination half-time of 76 h [71][72].
Galantamine (FDA 2001, Reminyl®) is a strictly a competitive inhibitor of AChE [75] that has an elimination half-time of 5–7 h [76]. It was likely the AChE inhibitor antidote in Homer’s Moly (Galanthus nivalis) that helped Odysseus rescue his crew from Circe’s malignant anticholinergic posset (Datura stramonium), which probably induced the central anticholinergic syndrome (stramonium poisoning), thousands of years ago [77]. Besides inhibiting AChE, galantamine is also an allosteric modulator of nicotinic acetylcholine receptors [78][79], which may lend it some clinical advantages [80].
3.1.2. Pseudo-Irreversible Inhibition: Rivastigmine and Metrifonate
Rivastigmine (FDA 2000, Exelon®) is classified as a pseudo-irreversible inhibitor because it reacts with the critical active site serine to form a covalent carbamoyl-AChE complex that precludes its catalysis of acetylcholine (Figure 1), but the inhibition is short-lived. The duration of rivastigmine-induced inhibition depends on the stability of that bond in the inactive carbamoyl-AChE complex [81]. Although the free drug molecule of rivastigmine is eliminated from the blood with a half-time of about 2.5 h, the covalent bond persists much longer, slowly undergoing spontaneous hydrolysis [81][82] so that rivastigmine-induced AChE inhibition persists for a period of ~8.5 h [71][72][83]. In addition, unlike donepezil and galantamine which are AChE-selective, rivastigmine also inhibits BChE with a duration of 3.5 h [72]. Inhibition of BChE has been proposed as an advantage depending on patient characteristics and genotype [84][85][86], especially for subcortical dementias [87], but BChE inhibition may also affect a range of non-neural functions and toxicities [88][89].
Metrifonate (BAY-A-9826, ProMem, 1997), an organophosphate, was introduced as an AChE inhibitor for the treatment of AD [90][91]. It has been described as a long-lasting cholinesterase inhibitor [82]. Metrifonate, introduced in humans as an acute treatment for schistosomiasis [92], undergoes in vivo spontaneous non-enzymatic rearrangement to 2,2-dicholorvinyldimethyl phosphate (DDVP, dichlorvos) [93][94] with a half-time of ~6 h at pH 7.0 [94][95]. Metrifonate and DDVP both inhibit BChE and AChE [96][97][98]. Most of the cholinesterase inhibition after in vivo administration is due to DDVP [94]. However, the phosphonyl-enzyme covalent bond between DDVP and the catalytic site of CNS AChE (Figure 1) in vivo undergoes spontaneous hydrolysis that results in a reactivated enzyme with a half-time of ~3–4 h [97][99]. Due to the ready hydrolysis of the covalent bond in vivo and resulting enzyme reactivation, metrifonate is most correctly characterized as a pseudo-irreversible inhibitor. Like other short-acting inhibitors, including rivastigmine, it showed little efficacy in treating dementia [90][91]. However, DDVP under its various names is well known to cause organophosphate-induced delayed neuropathy, a late-appearing toxicity that is not related to cholinesterase inhibition, [100][101][102][103][104] and is also a potent inhibitor of cytochrome oxidase [105]. Metrifonate was abandoned as a treatment for AD because it produces severe muscular and life-threatening respiratory paralysis in some AD patients, a sign of organophosphate-induced delayed neuropathy [91][103].
3.1.3. The Failure of Competitive and Pseudo-Irreversible Inhibitors
The fundamental and, so far, insurmountable problem with the current AChE inhibitors in either the clinical management or disease-modifying effects in AD is that there is no discoverable difference between the molecular architecture of CNS and peripheral AChE catalytic sites that has led to successfully identifying an inhibitor for CNS enzyme that does not also inhibit the peripheral enzyme. The result of this failure is that potent inhibition of CNS AChE invariably results in overstimulation of essential cholinomimetic mechanisms in peripheral tissues, especially gastrointestinal control which is highly sensitive to AChE-induced overstimulation. Overstimulation of the gastrointestinal tract causes intolerable dose-limiting nausea, vomiting, and diarrhea [70][71][72] and limits CNS AChE inhibition to the ineffective levels [65][66][67][68][69]. The current AChE inhibitors approved for the treatment of AD are not adequate for meaningful relief from AD-induced suffering or for useful medicoeconomic benefits [57][58][59][60][61].
Both clinical efficacy and adverse events induced by AChE inhibitors are dose-dependent [106], which indicates that high-level CNS AChE inhibition (above 50%) [52] will likely improve efficacy if the problem of adverse events can be overcome [52][107]. Increased CNS AChE inhibition, above what is currently available, will also improve the “CSF Cholinergic Index”, an in vivo physiological measure of an improved CNS ratio of AChE inhibition compared to increased choline acetyltransferase in AD patients [108]. However, high-level AChE inhibition (above the currently available inadequate clinical doses) is blocked by ubiquitous gastrointestinal toxicity produced by currently available AChE inhibitors [70][71][72]. High-level human CNS AChE inhibition (above 50%) in AD patients has only been available in one study that showed promising cognitive enhancement [109]. High-level CNS AChE inhibition in the treatment of AD is an important goal that deserves further study [52]. The single most important objective for the full realization of AChE inhibitor-induced cognitive improvements and anti-neurodegenerative benefits is obtaining effective CNS-selectivity [52][109][110]. The short-term competitive and pseudo-irreversible inhibitors have not been able to meet this fundamental requirement.
3.2. Mechanism of Action of Irreversible Inhibitors—CNS-Selectivity
3.2.1. Advantages of Irreversible AChE Inhibition
Irreversible inhibition differs from pseudo-irreversible inhibition in the stability of the covalent bond in the inhibitor-enzyme complex. In the case of pseudo-irreversible inhibition, the covalent bond in the inhibitor-enzyme complex is sufficiently weak so that it undergoes spontaneous hydrolysis, which results in complete reactivation of the enzyme to its full original capacity. In the case of truly irreversible inhibition, however, the covalent inhibitor-enzyme complex is sufficiently strong to be refractory to spontaneous hydrolysis and it permanently inactivates the enzyme molecule. The only way enzyme activity can be restored after irreversible inhibition is through de novo new synthesis of the enzyme. Thus, the duration of irreversible AChE inhibition depends on the rate at which new enzyme is being manufactured, the turnover rate, a characteristic of each tissue [110]. The only clinically useful difference between CNS and peripheral AChE to date is the discovery that CNS AChE is replaced at a much slower rate (t1/2 ~12 days) than in the peripheral tissues such as the smooth muscle of the gastrointestinal tract, cardiac muscle, and skeletal muscles (t1/2 as short as 1 day). This was recognized as an important tissue-specific difference that might be used, for the first time in the history of AD treatment, to produce CNS-selective AChE inhibition [110].
Figure 2 shows the magnitude of selectivity of an irreversible inhibitor toward CNS AChE inhibition that can be expected from very slow de novo enzyme replacement in the CNS (~12 days) versus fast replacement in peripheral tissues (~1 day). Figure 2 models drug administration given daily for 21 days to approximate a clinically relevant dose of an irreversible AChE inhibitor. These computations, explained in detail elsewhere [52], show that high AChE inhibition (~65%) is expected to accumulate in the CNS because of the slow recovery of activity between doses versus the low expected AChE inhibition (~20%) in peripheral tissues where much of the activity is replaced between doses. The large difference between the rate of de novo AChE replacement in the CNS and peripheral tissues is a key difference that can be exploited to produce highly selective CNS AChE inhibition.
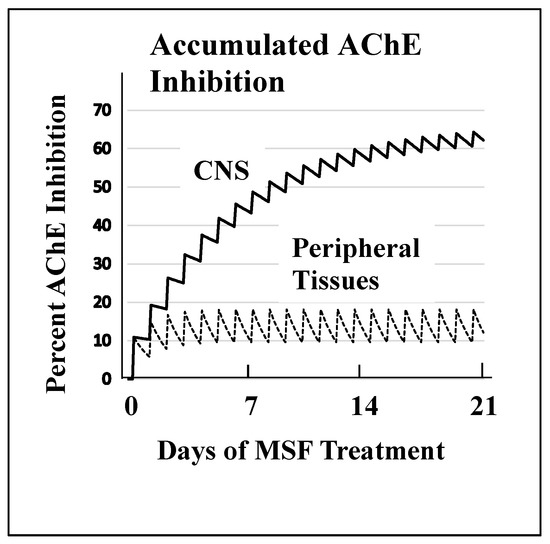
Figure 2. A computational model of the expected accumulated AChE inhibition in the CNS (upper solid line) versus peripheral tissues (lower dotted line) during three weeks of daily doses of an irreversible inhibitor (e.g., methanesulfonyl fluoride, MSF), computed as producing an equal 10% inhibition of currently active AChE in both CNS and peripheral tissues with each dose. The saw-tooth appearance of the lines shows the increment of inhibition (upward points) added with each dose. The downward slope between doses is the decrease in inhibition produced by new synthesis of the enzyme in the dose-to-dose interval. MSF disappears rapidly from blood, within a few hours, producing the pulsatile inhibition shown above. These pharmacological calculations (repeated dosing with recovery between doses) predict the accumulated effects occurring over 21 days [111]. The separation between the levels of CNS versus peripheral tissue accumulated AChE inhibition caused by differences in enzyme recovery rates, as shown above, does not occur with short-acting competitive or pseudo-irreversible inhibitors [52]. (Modified from Journal of Alzheimer’s Disease, 55, Cholinesterase Inhibitor Therapy in Alzheimer’s Disease: The Limits and Tolerability of Irreversible CNS-Selective Acetylcholinesterase, 1285–1294 (2017), with permission of IOS Press.The publication is available at IOS Press through http://dx.doi:10.3233/JAD-160733).The validity of the pharmacodynamics shown in Figure 2 was tested in an experiment in which rats were treated with methanesulfonyl fluoride (MSF), an irreversible AChE inhibitor, in accordance with the 21 day protocol modeled in Figure 2. In this experiment, which is explained in detail elsewhere [111], rats were sacrificed at the end of 21 days of treatment with MSF. As modeled by the computations, CNS AChE was inhibited much more (~75%) than AChE in peripheral tissues (<25% AChE), all without observable signs of toxicity (Figure 3). Seventy-five percent CNS AChE inhibition is at the upper end of the expected therapeutic window for AD and <25% is well below the beginning of toxicity from peripheral tissues [52][70][71][72]. Similarly, rats aged 24 months were pretreated with MSF in a computationally based 4 week protocol designed to produce ~50% CNS AChE inhibition, actually showed in 56% inhibition ex vivo, and such MSF pretreatment enhanced memory function in the aged animals to that equal to young animals [112]. The ability to produce highly selective CNS AChE inhibition without peripheral toxicity has been further confirmed in monkeys (Macaca fascicularis) treated with escalating doses of MSF over 3 months, ending with ten weeks of continuous MSF treatment at 5 times the human clinical dose. Cortical biopsies confirmed ~80% and ~45% cortical AChE and BChE inhibition, respectively, with no gastrointestinal toxicity, no neuropathy, nor any other troublesome effects [52].
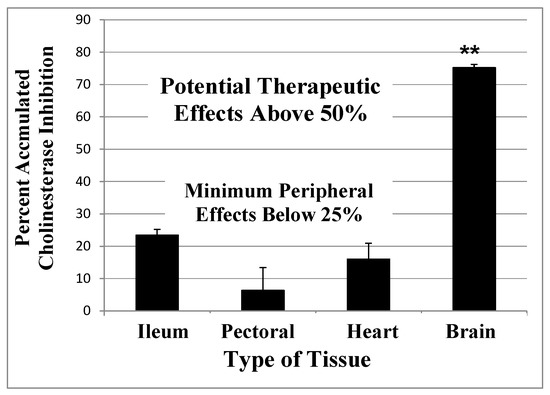
Figure 3. Accumulated AChE inhibition in four rat tissues after three weeks of repeated doses of 0.3 mg/kg MSF (IM) given three times per week to approximate the smaller daily dose shown in Figure 2. The animals were sacrificed 24 h after the last injection and smooth muscle (ileum), skeletal muscle (pectoral), cardiac muscle (heart), and whole brain were assayed for AChE inhibition, compared to untreated controls. CNS is significantly more inhibited than peripheral tissues (**p < 0.01), but peripheral tissues are not different from each other. Error bars show SEM [111]. (From British Journal of Clinical Pharmacology, 75, A Randomized Phase 1 Study of Methanesulfonyl Fluoride, an Irreversible Cholinesterase Inhibitor, for the Treatment of Alzheimer’s Disease, 1231-1239 (2013), with permission Wiley Press)
In summary, Figure 2 and Figure 3 show that an AChE inhibitor with an irreversible mechanism of action given repeatedly over a period of time, similar to a clinical protocol in AD treatment [109], can produce a level of AChE inhibition that is at least double the inadequate 25–35% CNS AChE inhibition observed with the short-acting inhibitors [65][66][67][68][69]. The robust difference between CNS and peripheral tissue AChE inhibition produced by an irreversible inhibitor depends entirely on the difference between the rate at which AChE is newly synthesized in the CNS as compared to peripheral tissues. High-level AChE inhibition can be produced and maintained in the CNS without gastrointestinal toxicity, but only if an irreversible inhibitor is used [52][109].
3.2.2. Sulfonyl Fluorides as AD Relevant Irreversible Inhibitors
Sulfonyl fluorides, including methanesulfonyl fluoride, have been known as irreversible AChE inhibitors since 1954 [113] with a well-understood and solidly irreversible mechanism of action that has been used as a molecular probe of the catalytic site of AChE since the early 1960s [114][115].
The sulfonyl fluorides, like carbamates (e.g., rivastigmine) and organophosphates [116], react covalently with the essential serine oxygen in the catalytic site of AChE to block the enzyme catalytic mechanism (Figure 1) [96][114][115]. The sulfonyl fluorides, specifically including MSF, do not inhibit neuropathy target enzyme, the hypothesized cause of organophosphate-induced delayed neuropathy [117]. Unlike the pseudo-irreversible inhibitors like rivastigmine and metrifonate, however, the sulfonyl-enzyme covalent complex is exceptionally stable and does not undergo spontaneous hydrolysis [114][115], nor can the enzyme be reactivated by strong oxime nucleophilic attack on the covalent bond [118]. Because there is no spontaneous reactivation of the enzyme, the irreversible sulfonyl-enzyme covalent complex was the first tool used to discover that the rate of de novo replacement of CNS AChE activity is more than 10× slower than AChE replacement in peripheral tissues in vivo [110]. Further study of the sulfonyl fluorides indicated that MSF, the smallest and most reactive of the sulfonyl fluorides, was ~100× more biologically active than the larger compounds [119], and the best candidate for the treatment of AD [120].
MSF has uncommon pharmacokinetics. Even though MSF-induced inhibition of CNS AChE disappears with a half-time of ~12 days, the time required for new synthesis, the MSF drug molecule itself is unstable in an aqueous environment such as human blood and undergoes inactivation by in vivo spontaneous hydrolysis to form methanesulfonic acid, an inactive compound, with a half-time of 2.6 h [111][116]. Therefore, MSF administered on a daily schedule like that simulated in Figure 2 produces a pulsatile increment in AChE inhibition that is followed by a drug-free period of ~16 h per day during which new synthesis of uninhibited replacement AChE occurs [52].
The use of MSF for the treatment of AD introduced a special problem. Insofar as MSF is highly selective for the CNS and is free from peripheral toxicity, the optimum dose for patients cannot be determined by increasing the dose until peripheral toxicity is observed, the procedure used for the short-acting AChE inhibitors [70][71][72]. Therefore, the first use of MSF in humans [109] required dose-estimation from the pharmacodynamics calculations shown in Figure 2 [111]. As predicted from the pharmacodynamics calculations and animal experiments (Figure 2 and Figure 3) [52][111], 8 weeks of oral MSF given three times per week to mild to moderate AD patients correctly produced an estimated ~66% CNS AChE inhibition [109], a level of CNS AChE inhibition that is at the upper end of the useful therapeutic window [70][71][72] and which resulted in strong cognitive improvement (~6 points on the ADAS-cog). Furthermore, the MSF-induced cognitive improvement persisted unabated through an additional 8 weeks of placebo [109].
After 8 weeks of placebo treatment, about 5 half-times for the de novo replacement of MSF-inhibited enzyme [52], only ~4% inhibition would remain. Therefore, the duration of strong cognitive improvement over 8 weeks, without further MSF treatment, suggests that MSF produced some long-term benefit that outlasted the direct effects of AChE inhibition. This contention is also supported by an experiment in which MSF treatment also preserves cholinergic neurons and choline acetyltransferase immunoreactivity in the basal forebrain of ischemic rats [121]. These data suggest that MSF-induced AChE inhibition has long-term disease-modifying benefits, perhaps by enhancing acetylcholine-dependent stimulation of NGF production and release and associated basal forebrain survival processes [2][46][47][48][49][50][51].
The high level of MSF-induced CNS AChE inhibition should be equaled by any truly irreversible inhibitor. The CNS selectivity of irreversible AChE inhibitors is due to the slow turnover rate of AChE in the CNS, not a property of the inhibitor molecule beyond the fact that it must form a sufficiently stable inhibitor-enzyme inactive complex that does not undergo spontaneous hydrolysis.
References
- P. Davies; SELECTIVE LOSS OF CENTRAL CHOLINERGIC NEURONS IN ALZHEIMER'S DISEASE. The Lancet 1976, 2, 1403, 10.1016/s0140-6736(76)91936-x.
- A. Claudio Cuello; Rowan Pentz; Hélène Hall; The Brain NGF Metabolic Pathway in Health and in Alzheimer's Pathology. Frontiers in Neuroscience 2019, 13, 62, 10.3389/fnins.2019.00062.
- Giancarlo Pepeu; Maria Grazia Giovannini; The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Research 2017, 1670, 173-184, 10.1016/j.brainres.2017.06.023.
- Nicolaas I Bohnen; Michel J. Grothe; Nicola J. Ray; Martijn Muller; Stefan J. Teipel; Recent Advances in Cholinergic Imaging and Cognitive Decline—Revisiting the Cholinergic Hypothesis of Dementia. Current Geriatrics Reports 2018, 7, 1-11, 10.1007/s13670-018-0234-4.
- Harald Hampel; M.-Marsel Mesulam; A Claudio Cuello; Martin R. Farlow; Ezio Giacobini; George T Grossberg; Ara S. Khachaturian; Andrea Vergallo; Enrica Cavedo; Peter J. Snyder; Zaven S. Khachaturian; The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917-1933, 10.1093/brain/awy132.
- Peter J. Whitehouse; Nald L. Price; Arthur W. Clark; Joseph T. Coyle; Mahlon R. Delong; Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Annals of Neurology 1981, 10, 122-126, 10.1002/ana.410100203.
- Hanna Al-Shaikh, F.S.; Duara, R.; Crook, J.E.; Lesser, E.R.; Schaeverbeke, J.; Hinkle, K.M.; Ross, O.A.; Ertekin-Taner, N.; Pedraza, O.; Dickson, D.W.; et al.et al Selective vulnerability of the nucleus basalis of Meynert among neuropathologic subtypes of Alzheimer disease. JAMA Neurol. 2019, 32224, 1–9.
- Taylor W Schmitz; R. Nathan Spreng; Michael W. Weiner; Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nature Communications 2016, 7, 13249, 10.1038/ncomms13249.
- Sara Fernández-Cabello; Martin Kronbichler; Koene R A Van Dijk; James A Goodman; R Nathan Spreng; Taylor W Schmitz; On Behalf Of The Alzheimer’S Disease Neuroimaging Initiative; Basal forebrain volume reliably predicts the cortical spread of Alzheimer’s degeneration. Brain 2020, 143, 993-1009, 10.1093/brain/awaa012.
- Stefan J. Teipel; Helmut Heinsen; Edson Amaro; Lea T. Grinberg; Bernd Krause; Michel J. Grothe; Alzheimer's Disease Neuroimaging Initiative; Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer's disease. Neurobiology of Aging 2014, 35, 482-491, 10.1016/j.neurobiolaging.2013.09.029.
- Taylor W Schmitz; Hermona Soreq; Judes Poirier; R. Nathan Spreng; For The Alzheimer's Disease Neuroimaging Initiative; Longitudinal Basal Forebrain Degeneration Interacts with TREM2/C3 Biomarkers of Inflammation in Presymptomatic Alzheimer's Disease. The Journal of Neuroscience 2020, 40, 1931-1942, 10.1523/jneurosci.1184-19.2019.
- M.-M Mesulam; Pamela Shaw; Deborah Mash; Sandra Weintraub; Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Annals of Neurology 2004, 55, 815-828, 10.1002/ana.20100.
- M.-M Mesulam; Cholinergic Circuitry of the Human Nucleus Basalis and Its Fate in Alzheimer's Disease. The Journal of Comparative Neurology 2013, 521, 4124-4144, 10.1002/cne.23415.
- I. Sassin; C Schultz; Dietmar Rudolf Thal; U. Rüb; K. Arai; E. Braak; H. Braak; Evolution of Alzheimer's disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathologica 2000, 100, 259-269, 10.1007/s004019900178.
- Chelsea T Tiernan; Elliott J Mufson; Nicholas M. Kanaan; Scott E. Counts; Tau Oligomer Pathology in Nucleus Basalis Neurons During the Progression of Alzheimer Disease. Journal of Neuropathology & Experimental Neurology 2018, 77, 246-259, 10.1093/jnen/nlx120.
- Andrew M. Hall; Robert Y. Moore; Oscar L. Lopez; Lewis Kuller; James T. Becker; Andrew M. Ho; Basal forebrain atrophy is a presymptomatic marker for Alzheimer's disease. Alzheimer's & Dementia 2008, 4, 271-279, 10.1016/j.jalz.2008.04.005.
- Bettina Laursen; Arne Mørk; Niels Plath; Uffe Kristiansen; Jesper Frank Bastlund; Cholinergic degeneration is associated with increased plaque deposition and cognitive impairment in APPswe/PS1dE9 mice. Behavioural Brain Research 2013, 240, 146-152, 10.1016/j.bbr.2012.11.012.
- Juan Jose Ramos-Rodriguez; Mar Pacheco-Herrero; Diana Thyssen; Maria Isabel Murillo-Carretero; Esther Berrocoso; Tara L. Spires-Jones; Brian J. Bacskai; Monica Garcia‐Alloza; Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice.. Journal of Neuropathology & Experimental Neurology 2013, 72, 272-285, 10.1097/NEN.0b013e318288a8dd.
- Wallace, W.; Ahlerst, S.T.; Gotlib, J.; Braginu, V.; Sugaro, J.; Gluck, R.; Sheat, P.A.; Davis, K.L.; Haroutunian, V. Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation (nucleus basalis of Meynert/rat). Neurobiol. Commun. 1993, 90, 8712–8716.
- Ilya D. Ionov; Irina I. Pushinskaya; Amyloid-β production in aged guinea pigs: atropine-induced enhancement is reversed by naloxone. Neuroscience Letters 2010, 480, 83-86, 10.1016/j.neulet.2010.06.010.
- T. Beach; D. Walker; L. Sue; S. Scott; K. Layne; A. Newell; P. Potter; R. Durham; M. Emmerling; S. Webster; Immunotoxin Lesion of the Cholinergic Nucleus Basalis Causes Aβ Deposition: Towards a Physiologic Animal Model of Alzheimers Disease. Current Medicinal Chemistry-Immunology, Endocrine & Metabolic Agents 2003, 3, 57-75, 10.2174/1568013033358635.
- D L Price; L J Martin; S S Sisodia; M V Wagster; E H Koo; L C Walker; V E Koliatsos; L C Cork; Aged non-human primates: an animal model of age-associated neurodegenerative disease. Brain Pathology 1991, 1, 287–296.
- Yasumasa Yoshiyama; Ayako Kojima; Chieko Ishikawa; Kimihito Arai; Anti-Inflammatory Action of Donepezil Ameliorates Tau Pathology, Synaptic Loss, and Neurodegeneration in a Tauopathy Mouse Model. Journal of Alzheimer's Disease 2010, 22, 295-306, 10.3233/jad-2010-100681.
- Elaine K. Perry; Linda Kilford; Andrew J. Lees; David Burn; Robert H. Perry; Increased Alzheimer pathology in Parkinson's disease related to antimuscarinic drugs. Annals of Neurology 2003, 54, 235-238, 10.1002/ana.10639.
- Shelly L. Gray; Melissa L. Anderson; Sascha Dublin; Joseph T. Hanlon; Rebecca Hubbard; Rod Walker; Onchee Yu; Paul K. Crane; Eric B. Larson; Cumulative use of strong anticholinergics and incident dementia: a prospective cohort study. JAMA Internal Medicine 2015, 75, 401-407, 10.1001/jamainternmed.2014.7663.
- Isabelle Carriere; Annie Fourrier-Réglat; Jean-François Dartigues; Olivier Rouaud; Florence Pasquier; Karen Ritchie; Marie-Laure Ancelin; Drugs With Anticholinergic Properties, Cognitive Decline, and Dementia in an Elderly General Population. Archives of Internal Medicine 2009, 169, 1317-24, 10.1001/archinternmed.2009.229.
- Risacher, S.L.; McDonald, B.; Tallman, E.; West, J.; Farlow, M.R.; Unverzagt, F.W.; Gao, S.; Boustani, M; Association Between Anticholinergic Medication Use and Cognition, Brain Metabolism, and Brain Atrophy in Cognitively Normal Older Adults. JAMA Neurology 2016, 73, 721-732, 10.1001/jamaneurol.2016.0580.
- Yi-Fang Chuang; Palchamy Elango; Christopher E. Gonzalez; Madhav Thambisetty; Midlife anticholinergic drug use, risk of Alzheimer's disease, and brain atrophy in community-dwelling older adults. Alzheimer's & Dementia: Translational Research & Clinical Interventions 2017, 3, 471-479, 10.1016/j.trci.2017.06.004.
- Alireza Atri; Lynn W. Shaughnessy; Joseph J. Locascio; John H. Growdon; Long-term Course and Effectiveness of Combination Therapy in Alzheimer Disease. Alzheimer Disease & Associated Disorders 2008, 22, 209-221, 10.1097/wad.0b013e31816653bc.
- Susan Rountree; Alireza Atri; Oscar L. Lopez; Rachelle S. Doody; Effectiveness of antidementia drugs in delaying Alzheimer's disease progression. Alzheimer's & Dementia 2013, 9, 338-345, 10.1016/j.jalz.2012.01.002.
- Oscar L. Lopez; J T Becker; Abdus Wahed; J Saxton; R A Sweet; D A Wolk; William Klunk; Steven T. DeKosky; Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. Journal of Neurology, Neurosurgery & Psychiatry 2009, 80, 600-607, 10.1136/jnnp.2008.158964.
- Carolyn W. Zhu; Elayne E. Livote; Nikolaos Scarmeas; Marilyn Albert; J. Brandt; Deborah Blacker; Mary Sano; Yaakov Stern; Long-term associations between cholinesterase inhibitors and memantine use and health outcomes among patients with Alzheimer's disease. Alzheimer's & Dementia 2013, 9, 733-740, 10.1016/j.jalz.2012.09.015.
- Elio Scarpini; Giuseppe Bruno; Giuseppe Zappalà; Marina Adami; Ute Richarz; Maren Gaudig; Adam Jacobs; Barbara Schäuble; Cessation versus Continuation of Galantamine Treatment after 12 Months of Therapy in Patients with Alzheimer's Disease: A Randomized, Double Blind, Placebo Controlled Withdrawal Trial. Journal of Alzheimer's Disease 2011, 26, 211-220, 10.3233/jad-2011-110134.
- Sean Lilienfeld; Wim Parys; Galantamine: additional benefits to patients with Alzheimer's disease. Dementia and Geriatric Cognitive Disorders 2000, 11, 19-27, 10.1159/000051228.
- R Blesa; Galantamine: therapeutic effects beyond cognition. Dementia and Geriatric Cognitive Disorders 2000, 11, 28-34, 10.1159/000051229.
- Agneta Nordberg; Taher Darreh-Shori; Elaine Peskind; Hilkka Soininen; Malahat Mousavi; Gina Eagle; Roger Lane; Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients.. Current Alzheimer Research 2009, 6, 4-14, 10.2174/156720509787313961.
- Ferris, S.; Nordberg, A.; Soininen, H.; Darreh-Shori, T.; Lane, R. Progression from mild cognitive impairment to Alzheimer’s disease: Effects of gender, butyrylcholinesterase genotype and rivastigmine treatment. Pharmacogenet Genomics. 2009, 19, 635–646.
- Annalena Venneri; Roger Lane; Effects of cholinesterase inhibition on brain white matter volume in Alzheimerʼs disease. NeuroReport 2009, 2, 285-288, 10.1097/wnr.0b013e3283207d21.
- Bo-Lin Ho; Yi-Hui Kao; Mei-Chuan Chou; Yuan-Han Yang; Cerebral White Matter Changes on Therapeutic Response to Rivastigmine in Alzheimer’s Disease. Journal of Alzheimer's Disease 2016, 54, 351-357, 10.3233/jad-160364.
- R. Douglas Fields; Dipankar Dutta; Jillian Belgrad; Maya Robnett; Cholinergic signaling in myelination. Glia 2017, 65, 687-698, 10.1002/glia.23101.
- Sultan Darvesh; Butyrylcholinesterase as a Diagnostic and Therapeutic Target for Alzheimer's Disease. Current Alzheimer Research 2016, 13, 1173–1177.
- Dubois, B.; Chupin, M.; Hampel, H.; Lista, S.; Cavedo, E.; Croisile, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; et al.et al Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer's disease. Alzheimer's & Dementia 2015, 11, 1041-1049, 10.1016/j.jalz.2014.10.003.
- Cavedo, E.; Dubois, B.; Colliot, O.; Lista, S.; Croisile, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; Rouaud, O.; et al.et al Reduced Regional Cortical Thickness Rate of Change in Donepezil-Treated Subjects With Suspected Prodromal Alzheimer’s Disease. The Journal of Clinical Psychiatry 2016, 77, e1631-e1638, 10.4088/JCP.15m10413.
- Enrica Cavedo; Hippocampus Study Group; Michel J. Grothe; Olivier Colliot; Simone Lista; Marie Chupin; Didier Dormont; Marion Houot; Stephane Lehéricy; Stefan Teipel; Bruno Dubois; Harald Hampel; Bernard Croisile; Reduced basal forebrain atrophy progression in a randomized Donepezil trial in prodromal Alzheimer’s disease. Scientific Reports 2017, 7, 11706, 10.1038/s41598-017-09780-3.
- K. R. R. Krishnan; H. Cecil Charles; P. Murali Doraiswamy; Jacobo Mintzer; Richard Weisler; Xin Yu; Carlos Perdomo; John R. Ieni; Sharon Rogers; Randomized, Placebo-Controlled Trial of the Effects of Donepezil on Neuronal Markers and Hippocampal Volumes in Alzheimer’s Disease. American Journal of Psychiatry 2003, 160, 2003-2011, 10.1176/appi.ajp.160.11.2003.
- Martin A. Bruno; A. Claudio Cuello; Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proceedings of the National Academy of Sciences 2006, 103, 6735-6740, 10.1073/pnas.0510645103.
- A. Claudio Cuello; Martin A. Bruno; Simon Allard; Wanda Leon; M. Florencia Iulita; Cholinergic Involvement in Alzheimer’s Disease. A Link with NGF Maturation and Degradation. Journal of Molecular Neuroscience 2010, 40, 230-235, 10.1007/s12031-009-9238-z.
- Xu-Qiao Chen; William C. Mobley; Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses. Frontiers in Neuroscience 2019, 13, 446, 10.3389/fnins.2019.00446.
- Margaret Fahnestock; Arman Shekari; ProNGF and Neurodegeneration in Alzheimer's Disease. Frontiers in Neuroscience 2019, 13, 129, 10.3389/fnins.2019.00129.
- Valentina Latina; Silvia Caioli; Cristina Zona; Maria T. Ciotti; Giuseppina Amadoro; Pietro Calissano; Impaired NGF/TrkA Signaling Causes Early AD-Linked Presynaptic Dysfunction in Cholinergic Primary Neurons. Frontiers in Cellular Neuroscience 2017, 11, 1-23, 10.3389/fncel.2017.00068.
- Scott E. Counts; Elliott J. Mufson; The Role of Nerve Growth Factor Receptors in Cholinergic Basal Forebrain Degeneration in Prodromal Alzheimer Disease. Journal of Neuropathology & Experimental Neurology 2005, 64, 263-272, 10.1093/jnen/64.4.263.
- Donald E. Moss; Ruth G. Perez; Haruo Kobayashi; Cholinesterase Inhibitor Therapy in Alzheimer’s Disease: The Limits and Tolerability of Irreversible CNS-Selective Acetylcholinesterase Inhibition in Primates. Journal of Alzheimer's Disease 2017, 55, 1285-1294, 10.3233/jad-160733.
- J. A. Deutsch; The Cholinergic Synapse and the Site of Memory. Science 1971, 174, 788-794, 10.1126/science.174.4011.788.
- Monica Janeczek; Tamar Gefen; Mehrnoosh Samimi; Garam Kim; Sandra Weintraub; Eileen Bigio; Emily Rogalski; M -Marsel Mesulam; Changiz Geula; Variations in Acetylcholinesterase Activity within Human Cortical Pyramidal Neurons Across Age and Cognitive Trajectories. Cerebral Cortex 2018, 28, 1329-1337, 10.1093/cercor/bhx047.
- Bartus, R.T.; Dean, R.L.; Pontecorvo, M.J.; Flicker, C. The cholinergic hypothesis: A historical overview, current perspective, and future irections. Ann. N. Y. Acad. Sci. 1985, 444, 332–358.
- D A Drachman; J Leavitt; Human memory and the cholinergic system. A relationship to aging?. Archives of Neurology 1974, 30, 113–127.
- Bond, M.; Rogers, G.; Peters, J.; Anderson, R.; Hoyle, M.; Miners, A.; Moxham, T.; Davis, S.; Thokala, P.; Wailoo, A.; et al.et al The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): a systematic review and economic model. Health Technology Assessment 2012, 16, 1-470, 10.3310/hta16210.
- Howard H. Feldman; Tuula Pirttilä; Jean François Dartigues; Barry John Everitt; Bart Van Baelen; Susanne Schwalen; Shane Kavanagh; Treatment with galantamine and time to nursing home placement in Alzheimer's disease patients with and without cerebrovascular disease. International Journal of Geriatric Psychiatry 2009, 24, 479-488, 10.1002/gps.2141.
- Hommet, C.; Novella, J.; Auriacombe, S.; Vercelletto, M.; Berrut, G.; Belliard, S.; Desmidt, T.; Ceccaldi, M.; Centre, C.; Tours, C. Les traitements symptomatiques à partir des Centres mémoire ressources. Geriatr. Psychol. Neuropsychiatr. du Vieil. 2016, 14, 274–286.
- Pierre Krolak-Salmon; Bruno Dubois; François Sellal; Jean-Philippe Delabrousse-Mayoux; Pierre Vandel; Hélène Amieva; Claude Jeandel; Sandrine Andrieu; Armand Perret-Liaudet; France Will No More Reimburse Available Symptomatic Drugs Against Alzheimer’s Disease. Journal of Alzheimer's Disease 2018, 66, 425-427, 10.3233/jad-180843.
- Emma Loveman; C Green; J Kirby; A Takeda; Joanna Picot; E Payne; A Clegg; Liz Payne; The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer's disease. Health Technology Assessment 2006, 10, 1–160, 10.3310/hta10010.
- Filip Zemek; Lucie Drtinova; Eugenie Nepovimova; Vendula Hepnarova; Jan Korábečný; Jiri Klimes; Kamil Kuca; Outcomes of Alzheimer's disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opinion on Drug Safety 2014, 13, 759–774, 10.1517/14740338.2014.914168.
- Js Birks; R Harvey; Donepezil for dementia due to Alzheimer’s disease. Cochrane Database of Systematic Reviews 2018, 6, CD001190, 10.1002/14651858.cd001190.
- Daniela Galimberti; Elio Scarpini; Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opinion on Investigational Drugs 2016, 25, 1181-1187, 10.1080/13543784.2016.1216972.
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.J.; Koeppe, R.A.; Meltzer, C.C.; Constantine, G.; Davis, J.G.; Mathis, C.A.; et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzeimer’s disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 315–319.
- David E. Kuhl; Satoshi Minoshima; Kirk A. Frey; Norman L. Foster; Michael R. Kilbourn; Robert A. Koeppe; Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Annals of Neurology 2000, 48, 391-395, 10.1002/1531-8249(200009)48:3<391::aid-ana17>3.0.co;2-h.
- Tsuneyoshi Ota; Hitoshi Shinotoh; Kiyoshi Fukushi; Tatsuya Kikuchi; Koichi Sato; Noriko Tanaka; Hitoshi Shimada; Shigeki Hirano; Michie Miyoshi; Heii Arai; Tetsuya Suhara; Toshiaki Irie; Estimation of Plasma IC50 of Donepezil for Cerebral Acetylcholinesterase Inhibition in Patients With Alzheimer Disease Using Positron Emission Tomography. Clinical Neuropharmacology 2010, 33, 74-78, 10.1097/wnf.0b013e3181c71be9.
- Valtteri Kaasinen Md; Kjell Någren; Tarja Järvenpää; Anne Roivainen; Meixiang Yu; Vesa Oikonen; Timo Kurki; Juha O. Rinne; Regional Effects of Donepezil and Rivastigmine on Cortical Acetylcholinesterase Activity in Alzheimer’s Disease. Journal of Clinical Psychopharmacology 2002, 22, 615-620, 10.1097/00004714-200212000-00012.
- A. Kadir; Taher Darreh-Shori; Ove Almkvist; A. Wall; M. Grut; B. Strandberg; Anna Ringheim; B. Eriksson; G. Blomquist; B. Långström; Agneta Nordberg; PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiology of Aging 2008, 29, 1204-1217, 10.1016/j.neurobiolaging.2007.02.020.
- Bruno Pietro Imbimbo; Pharmacodynamic-Tolerability Relationships of Cholinesterase Inhibitors for Alzheimer??s Disease. CNS Drugs 2001, 15, 375-390, 10.2165/00023210-200115050-00004.
- Jann, M.W.; Shirley, K.L.; Small, G.W; Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739.
- Muriel Noetzli; Chin B. Eap; Pharmacodynamic, Pharmacokinetic and Pharmacogenetic Aspects of Drugs Used in the Treatment of Alzheimer’s Disease. Clinical Pharmacokinetics 2013, 52, 225-241, 10.1007/s40262-013-0038-9.
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307.
- Hachiro Sugimoto; Youichi Iimura; Yoshiharu Yamanishi; Kiyomi Yamatsu; Synthesis and Structure-Activity Relationships of Acetylcholinesterase Inhibitors: 1-Benzyl-4-[(5,6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine Hydrochloride and Related Compounds. Journal of Medicinal Chemistry 1995, 38, 4821-4829, 10.1021/jm00024a009.
- T. Thomsen; H. Kewitz; Selective inhibition of human acetylcholinesterase by galanthamine in vitro and in vivo. Life Sciences 1990, 46, 1553-1558, 10.1016/0024-3205(90)90429-u.
- G. S. J. Mannens; C. A. W. Snel; J. Hendrickx; T. Verhaeghe; L. Le Jeune; W. Bode; L. Van Beijsterveldt; K. Lavrijsen; J. Leempoels; N. Van Osselaer; et al.A. Van PeerW. Meuldermans The metabolism and excretion of galantamine in rats, dogs, and humans. Drug Metabolism and Disposition 2002, 30, 553-563, 10.1124/dmd.30.5.553.
- A Plaitakis; R C Duvoisin; Homer's moly identified as Galanthus nivalis L.: physiologic antidote to stramonium poisoning. Clin. Neuropharmacol. 1983, 6, 1-5.
- M Samochocki; M Zerlin; R Jostock; P J Groot Kormelink; W H Luyten; E X Albuquerque; Alfred Maelicke; Galantamine is an allosterically potentiating ligand of the human alpha4/beta2 nAChR. Acta neurologica Scandinavica. Supplementum 2000, 176, 68–73.
- A Schrattenholz; E F Pereira; U Roth; K H Weber; E X Albuquerque; Alfred Maelicke; Agonist responses of neuronal nicotinic acetylcholine receptors are potentiated by a novel class of allosterically acting ligands. Molecular Pharmacology 1996, 49, 1-6.
- Sean Lilienfeld; Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer's disease. CNS Drug Reviews 2002, 8, 159–176.
- P. Bar-On; C. B. Millard; M. Harel; H. Dvir; A. Enz; Joel L. Sussman; I. Silman; Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine†,‡. Biochemistry 2002, 41, 3555-3564, 10.1021/bi020016x.
- Enz, A.; Floersheim, P. Cholinesterase inhibitors: An overview of their mechanisms of action. In Alzheimer’s Disease. Therapeutic Strategies; Giacobini, E., Becker, R., Eds.; Birkhauser: Boston, MA, USA, 1994; pp. 211–215.
- Roger M. Lane; Taher Darreh-Shori; Understanding the Beneficial and Detrimental Effects of Donepezil and Rivastigmine to Improve their Therapeutic Value. Journal of Alzheimer's Disease 2015, 44, 1039-1062, 10.3233/jad-142268.
- Roger M. Lane; Albert Enz; Steven Potkin; Targeting acetylcholinesterase and butyrylcholinesterase in dementia. The International Journal of Neuropsychopharmacology 2005, 9, 101–124, 10.1017/S1461145705005833.
- Agneta Nordberg; Clive Ballard; Roger Bullock; Taher Darreh-Shori; Monique Somogyi; A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. The Primary Care Companion For CNS Disorders 2013, 15, PCC.12r01412, 10.4088/PCC.12r01412.
- Dane R. Liston; Jann A. Nielsen; Anabella Villalobos; Douglas Chapin; Shawn B. Jones; Sean T. Hubbard; Ismail A. Shalaby; Andres Ramirez; Deane Nason; W Frost White; Pharmacology of selective acetylcholinesterase inhibitors: implications for use in Alzheimer's disease.. European Journal of Pharmacology 2004, 486, 9-17, 10.1016/j.ejphar.2003.11.080.
- Nagaendran Kandiah; Ming-Chyi Pai; Vorapun Senanarong; Irene Looi; Encarnita Ampil; Kyung Won Park; Ananda Krishna Karanam; Stephen Christopher; Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clinical Interventions in Aging 2017, 12, 697-707, 10.2147/CIA.S129145.
- Carey Pope; Stephen Brimijoin; Cholinesterases and the fine line between poison and remedy. Biochemical Pharmacology 2018, 153, 205-216, 10.1016/j.bcp.2018.01.044.
- Sultan Darvesh; David A. Hopkins; Changiz Geula; Neurobiology of butyrylcholinesterase. Nature Reviews Neuroscience 2003, 4, 131-138, 10.1038/nrn1035.
- Robert E. Becker; Jerry A Colliver; Stephen J. Markwell; Pamela L. Moriearty; Latha K. Unni; Sandra Vicari; Double-Blind, Placebo-Controlled Study of Metrifonate, an Acetylcholinesterase Inhibitor, for Alzheimer Disease. Alzheimer Disease & Associated Disorders 1996, 10, 124-131, 10.1097/00002093-199601030-00003.
- López-Arrieta, J.M.; Schneider, L. Metrifonate for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, CD003155.
- J. M. Jewsbury; M.J. Cooke; M.C. Weber; Field trial of metrifonate in the treatment and prevention of Schistosomiasis infection in man. Annals of Tropical Medicine & Parasitology 1977, 71, 67-83, 10.1080/00034983.1977.11687163.
- Ingrid Nordgren; Bo Holmstedt; Marianne Sandoz; Transformation and action of metrifonate. Archives of Toxicology 1978, 41, 31-41, 10.1007/bf00351767.
- Holmstedt, B.; Nordgren, I.; Sandoz, M.; Sundwall, A; Metrifonate. Arch. Toxicol. 1978, 41, 3–29, 10.1016/b978-008055232-3.62176-2.
- R. L. Metcalf; T. R. Fukuto; R. B. March; Toxic Action of Dipterex and DDVP to the House Fly. Journal of Economic Entomology 1959, 52, 44-49, 10.1093/jee/52.1.44.
- G Pacheco; R Palacios-Esquivel; D E Moss; Cholinesterase inhibitors proposed for treating dementia in Alzheimer’s disease: Selectivity toward human brain acetylcholinesterase compared with butyrylcholinesterase. Journal of Pharmacology and Experimental Therapeutics 1995, 74, 767–770.
- Haruo Kobayashi; Takuma Nakano; E.Moss Donald; Tadahiko Suzuki; Effects of a Central Anticholinesterase, Methanesulfonyl Fluoride on The Cerebral Cholinergic System and Behavior in Mice : Comparison with an Organophosphate DDVP. JOURNAL OF HEALTH SCIENCE 1999, 45, 191-202, 10.1248/jhs.45.191.
- L K Unni; C Womack; M E Hannant; R E Becker; Pharmacokinetics and pharmacodynamics of metrifonate in humans. Methods and Findings in Experimental and Clinical Pharmacology 1994, 16, 285–289.
- M Hallak; E Giacobini; A comparison of the effects of two inhibitors on brain cholinesterase. Neuropharmacology 1987, 26, 521–530.
- Zalewska, Z.; Rakowska, I.; Matraszek, G.; Sitkiewicz, D. Effect of dichlorvos on some enzymes activites of the rat brain during postnatal development. Neuropatol. Pol. 1977, 15, 255–262.
- Caroldi, S.; Lotti, M. Delayed neurotoxicity caused by a single masssive dose of dichlorvos to adult hens. Toxicol. Lett. 1981, 9, 157–159.
- C Vasilescu; A Florescu; Clinical and electrophysiological study of neuropathy after organophosphorus compounds poisoning. Archives of Toxicology 1980, 43, 305–315.
- Desi, I.; Nagymajtenyi, L. Neurotoxicologic investigations of the pesticide dichlorvos (DDVP): Effects on the central and peripheral nervous system. Toxicology 1988, 49, 141–148.
- Sitkiewicz, D.; Zalewska, Z. Aktywność oksydazy cytochromowej i dehydrogenazy bursztynianowej mózgu szczura po zatruciu fosforoorganicznymi insektycydami dichlorfosem i trichlorfonem [The activity of cytochrome oxidase and succinate dehydrogenase in rat brain mitochondria following trichlorphon and dichlorvos intoxication]. Neuropathol. Pol. 1975, 13, 279–280.
- Marcello Lotti; Promotion of organophosphate induced delayed polyneuropathy by certain esterase inhibitors. Toxicology 2002, 181–182, 245-248, 10.1016/s0300-483x(02)00291-3.
- Kai-Xin Dou; Meng-Shan Tan; Chen-Chen Tan; Xi-Peng Cao; Xiao-He Hou; Qi-Hao Guo; Lan Tan; Vincent Chung-Tong Mok; Jin-Tai Yu; Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: a network meta-analysis of 41 randomized controlled trials. Alzheimer's Research & Therapy 2018, 10, 126, 10.1186/s13195-018-0457-9.
- Thomas Chase; Martin R. Farlow; Kathleen Clarence-Smith; Donepezil Plus Solifenacin (CPC-201) Treatment for Alzheimer's Disease.. Neurotherapeutics 2017, 14, 405-416, 10.1007/s13311-016-0511-x.
- Azadeh Karami; Maria Eriksdotter; Ahmadul Kadir; Ove Almkvist; Agneta Nordberg; Taher Darreh-Shori; CSF Cholinergic Index, a New Biomeasure of Treatment Effect in Patients With Alzheimer's Disease. Frontiers in Neuroscience 2019, 12, 239, 10.3389/fnmol.2019.00239.
- Moss, D.E.; Berlanga, P.; Hagan, M.M.; Sandoval, H.; Ishida, C. Methanesulfonyl fluoride (MSF): A double-blind, placebo-controlled study of safety and efficacy in the treatment of senile dementia of the Alzheimer type. Alzheimer Dis. Assoc. Disord. 1999, 13, 20–25.
- Moss, D.E.; Rodriguez, L.; Selim, S.; Ellett, S.; Devine, J.; Steger, R. The sulfonyl fluorides: CNS selective cholinesterase inhibitors with potential value in Alzheimer’s disease? In Neurology and Neurobiology 18: Senile Dementia of the Alzheimer Type; Hutton, J.T., Kenny, A.D., Eds.; Alan, R. Liss: New York, NY, USA, 1985; pp. 337–350.
- Donald E. Moss; Ruggero G. Fariello; Jörg Sahlmann; Isabel Sumaya; Federica Pericle; Enrico Braglia; A randomized phase I study of methanesulfonyl fluoride, an irreversible cholinesterase inhibitor, for the treatment of Alzheimer's disease. British Journal of Clinical Pharmacology 2013, 75, 1231-1239, 10.1111/bcp.12018.
- David H. Malin; Robert E. Plotner; Sarah J. Radulescu; Robert N. Ferebee; J.Ronald Lake; Pilar G. Negrete; Peggy J. Schaefer; Marie K. Crothers; Donald E. Moss; Chronic methanesulfonyl fluoride enhances one-trial per day reward learning in aged rats. Neurobiology of Aging 1993, 14, 393-395, 10.1016/0197-4580(93)90127-w.
- Myers, D.; Kemp, A; Inhibition of esterases by the fluorides of organic acids. Nature 1954, 173, 33–34.
- R Kitz; I B Wilson; Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. Journal of Biological Chemistry 1962, 237, 3245–3249.
- Fahrney, D.; Gold, A; Sulfonyl fluorides as inhibitors of esterases. J. Am. Chem. Soc. 1963, 85, 997–1000.
- Arthur W. Snow; William R. Barger; A chemical comparison of methanesulfonyl fluoride with organofluorophosphorus ester anticholinesterase compounds. Chemical Research in Toxicology 1988, 1, 379-384, 10.1021/tx00006a009.
- Sulfonyl Fluorides and the Promotion of Diisopropyl Fluorophosphate Neuropathy. Toxicological Sciences 1996, 33, 294-297, 10.1093/toxsci/33.2.294.
- Moss, D.; Keathley, S. Pilot Study to Test Sulfnates’ Ability to Provide Prophylaxis Against Nerve Agents; Technical Report; (Contract No. DAMD 17-87-C-7064); The U.S. Army Medical Research and Development Command: Frederick, MD, USA, 1 July 1988.
- Moss, D.; Rodriguez, L.; Herndon, W.; Vincenti, S.; Camarena, M. Sulfonyl fluorides as possible therapeutic agents in Alzheimer’s disease: Structure/activity relationships as CNS selective cholinesterase inhibitors. In Alzheimer’s and Parkinson’s Disease: Strategies in Research and Development; Fisher, A., Lachman, C., Hanin, I., Eds.; Plenum Press: New York, NY, USA, 1986; pp. 551–556
- R L Palacios-Esquivel; G Pacheco; D E Moss; Methanesulfonyl fluoride (MSF) blocks scopolamine-induced amnesia in rats.. Neurobiology of Aging 1993, 14, , null.
- Cesar V. Borlongan; Isabel C. Sumaya; Donald E. Moss; Methanesulfonyl fluoride, an acetylcholinesterase inhibitor, attenuates simple learning and memory deficits in ischemic rats. Brain Research 2005, 1038, 50-58, 10.1016/j.brainres.2005.01.028.


