Acetylcholinesterase (AChE) inhibitors are the only drugs that have demonstrated success in slowing shrinkage (atrophy) of the cortex, hippocampus, and basal forebrain, major areas of Alzheimer's disease (AD)-associated brain damage and dementia. The main barrier to taking advantage of this new success in treating, or even preventing, AD is that the old available AChE inhibitors are weak reversible inhibitors that cause intolerable nausea, vomiting, and diarrhea if given in the higher doses needed. A promising new strategy for producing high-level AChE inhibition in the brain as needed for effective treatment of AD is the use of AChE inhibitors that are of a different type, the irreversible inhibitors.
- Acetylcholinesterase inhibitor
- Alzheimer's
- irreversible inhibitor
- prophylaxis
AChE Inhibitors and Anticholinergics Affect Neurodegeneration in AD
Failures of Current AChE Inhibitors
Mechanisms of Action of Key AChE Inhibitors
| Inhibitor | Mechanism of Action | Additional Notes * |
|---|---|---|
| Donepezil | Competitive/Noncompetitive | |
| Galantamine | Competitive | Upregulates nicotinic receptors |
| Rivastigmine | Pseudo-Irreversible | Also inhibits BChE |
| Metrifonate | Pseudo-Irreversible | Induces peripheral neuropathy |
| Methanesulfonyl Fluoride | Irreversible | High CNS Selectivity |
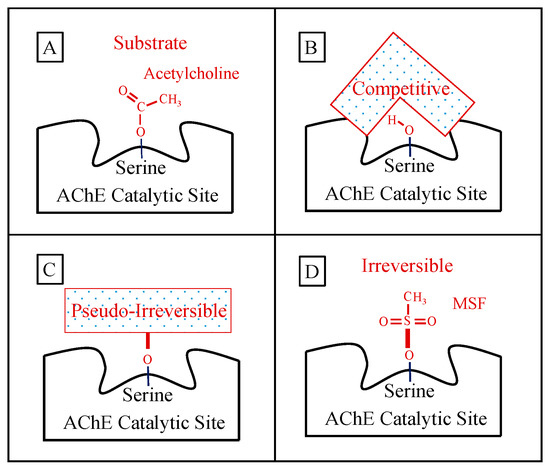
Mechanisms of Action of Short-Acting AChE Inhibitors—Limited Efficacy
Competitive AChE Inhibition: Donepezil and Galantamine
Pseudo-Irreversible Inhibition: Rivastigmine and Metrifonate
The Failure of Competitive and Pseudo-Irreversible Inhibitors
Mechanism of Action of Irreversible Inhibitors—CNS-Selectivity
Advantages of Irreversible AChE Inhibition
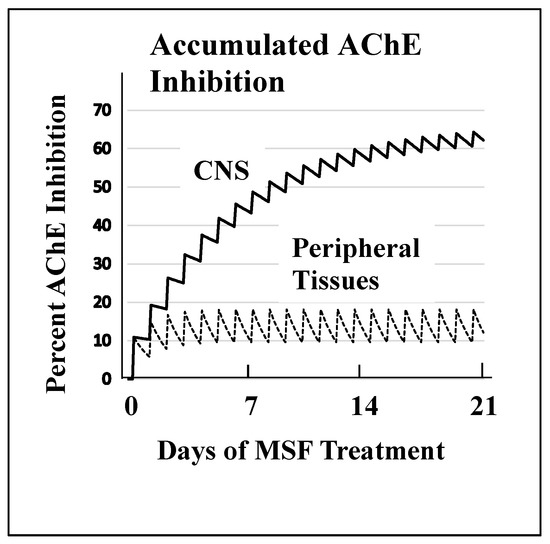
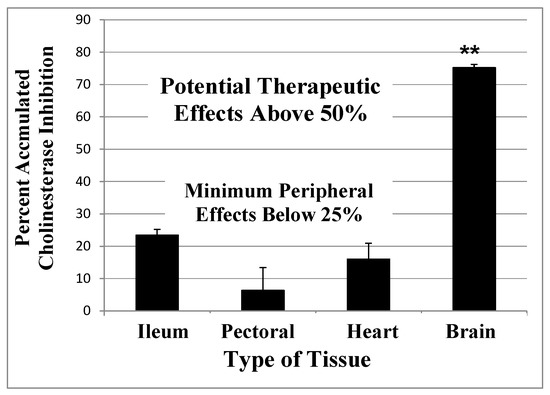
Sulfonyl Fluorides as AD Relevant Irreversible Inhibitors
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Cuello, C.A.; Pentz, R.; Hall, H. The brain NGF metabolic pathway in health and in Alzheimer’s pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannini, G.M. The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res. 2017, 1670, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Grothe, M.J.; Ray, N.J.; Müller, M.L.T.M.; Teipel, S.J. Recent advances in cholinergic imaging and cognitive decline—Revisiting the cholinergic hypothesis of dementia. Curr. Geriatr. Reports 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Hanna Al-Shaikh, F.S.; Duara, R.; Crook, J.E.; Lesser, E.R.; Schaeverbeke, J.; Hinkle, K.M.; Ross, O.A.; Ertekin-Taner, N.; Pedraza, O.; Dickson, D.W.; et al. Selective vulnerability of the nucleus basalis of Meynert among neuropathologic subtypes of Alzheimer disease. JAMA Neurol. 2019, 32224, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.W.; Spreng, R.N. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef]
- Fernández-Cabello, S.; Kronbichler, M.; Van Dijk, K.R.A.; Goodman, J.A.; Spreng, R.N.; Schmitz, T.W. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain volume reliably predicts the cortical spread of Alzheimer’s degeneration. Brain 2020, 143, 993–1009. [Google Scholar] [CrossRef]
- Teipel, S.; Heinsen, H.; Amaro, E.; Grinberg, L.T.; Krause, B.; Grothe, M. Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 482–491. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Soreq, H.; Poirier, J.; Spreng, R.N. Longitudinal basal forebrain degeneration interacts with TREM2/C3 biomarkers of inflammation in presymptomatic Alzheimer’s disease. J. Neurosci. 2020, 40, 1931–1942. [Google Scholar] [CrossRef]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 2004, 55, 815–828. [Google Scholar] [CrossRef]
- Mesulam, M.-M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef]
- Sassin, I.; Schultz, C.; Thal, D.R.; Rüb, U.; Arai, K.; Braak, E.; Braak, H. Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 2000, 100, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Tiernan, C.T.; Mufson, E.J.; Kanaan, N.M.; Counts, S.E. Tau oligomer pathology in nucleus basalis neurons during the progression of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2018, 77, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Moore, R.Y.; Lopez, O.L.; Kuller, L.; Becker, J.T. Basal forebrain atrophy is a presymptomatic marker for Alzheimer’s disease. Alzheimer’s Dement. 2008, 4, 271–279. [Google Scholar] [CrossRef]
- Laursen, B.; Mørk, A.; Plath, N.; Kristiansen, U.; Bastlund, J.F. Cholinergic degeneration is associated with increased plaque deposition and cognitive impairment in APPswe/PS1dE9 mice. Behav Brain Res. 2013, 240, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.; Ahlerst, S.T.; Gotlib, J.; Braginu, V.; Sugaro, J.; Gluck, R.; Sheat, P.A.; Davis, K.L.; Haroutunian, V. Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation (nucleus basalis of Meynert/rat). Neurobiol. Commun. 1993, 90, 8712–8716. [Google Scholar]
- Ionov, I.D.; Pushinskaya, I.I. Amyloid-β production in aged guinea pigs: Atropine-induced enhancement is reversed by naloxone. Neurosci. Lett. 2010, 480, 83–86. [Google Scholar] [CrossRef]
- Beach, T.G.; Walker, D.G.; Sue, L.I.; Scott, S.; Layne, K.J.; Newell, A.J.; Potter, P.E.; Durham, R.A.; Emmerling, M.R.; Webster, S.D. Immunotoxin lesion of the cholinergic nucleus basalis causes Aβ deposition: Towards a physiologic animal model of Alzheimer’s disease. Curr. Med. Chem. Imun., Endoc. Metab. Agents 2003, 3, 57–75. [Google Scholar] [CrossRef]
- Price, D.L.; Martin, L.J.; Sisodia, S.S.; Wagster, M.V.; Koo, E.H.; Walker, L.C.; Koliatsos, V.E.; Cork, L.C. Aged Non-Human Primates: An Animal Model of Age-Associated Neurodegenerative Disease. Brain Pathol. 1991, 1, 287–296. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Kojima, A.; Ishikawa, C.; Arai, K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J. Alzheimer’s Dis. 2010, 22, 295–306. [Google Scholar] [CrossRef]
- Perry, E.K.; Kilford, L.; Lees, A.J.; Burn, D.J.; Perry, R.H. Increased Alzheimer pathology in Parkinson’s disease related to antimuscarinic drugs. Ann. Neurol. 2003, 54, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Anderson, M.L.; Dublin, S.; Hanlon, J.T.; Hubbard, R.; Walker, R.; Yu, O.; Crane, P.K.; Larson, E.B. Cumulative use of strong anticholinergics and incident dementia: A prospective cohort study. JAMA Intern. Med. 2015, 75, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Carrière, I.; Fourrier-Reglat, A.; Dartigues, J.F.; Rouaud, O.; Pasquier, F.; Ritchie, K.; Ancelin, M.L. Drugs with anticholinergic properties, cognitive decline, and dementia in an elderly general population: The 3-city study. Arch. Intern. Med. 2009, 169, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; McDonald, B.; Tallman, E.; West, J.; Farlow, M.R.; Unverzagt, F.W.; Gao, S.; Boustani, M. Association between anticholinergic medication use and cognition, brain metabolism, and brain atrophy in cognitively normal older adults. JAMA Neurol. 2016, 73, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.; Elango, P.; Gonzalez, C.E.; Thambisetty, M. Midlife anticholinergic drug use, risk of Alzheimer’s disease, and brain atrophy in community-dwelling older adults. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Shaughnessy, L.W.; Locascio, J.J.; Growdon, J.H. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 209–221. [Google Scholar] [CrossRef]
- Rountree, S.D.; Atri, A.; Lopez, O.L.; Doody, R.S. Effectiveness of antidementia drugs in delaying Alzheimer’s disease progression. Alzheimer’s Dement. 2013, 9, 338–345. [Google Scholar] [CrossRef]
- Lopez, O.L.; Becker, J.T.; Wahed, A.S.; Saxton, J.; Sweet, R.A.; Wolk, D.A.; Klunk, W.; DeKosky, S.T. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 600–607. [Google Scholar] [CrossRef]
- Zhu, C.W.; Livote, E.E.; Scarmeas, N.; Albert, M.; Brandt, J.; Blacker, D.; Sano, M.; Stern, Y. Long-term associations between cholinesterase inhibitors and memantine use and health outcomes among patients with Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 733–740. [Google Scholar] [CrossRef]
- Scarpini, E.; Bruno, G.; Zappalà, G.; Adami, M.; Richarz, U.; Gaudig, M.; Jacobs, A.; Schäuble, B. Cessation versus continuation of galantamine treatment after 12 months of therapy in patients with alzheimer’s disease: A randomized, double blind, placebo controlled withdrawal trial. J. Alzheimer’s Dis. 2011, 26, 211–220. [Google Scholar] [CrossRef]
- Lilienfeld, S.; Parys, W. Galantamine: Additional benefits to patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2000, 11 (Suppl. S1), 19–27. [Google Scholar] [CrossRef]
- Blesa, R. Galantamine: Therapeutic effects beyond cognition. Dement Geriatr Cogn Disord. 2000, 11 (Suppl. S1), 28–34. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Darreh-Shori, T.; Peskind, E.; Soininen, H.; Mousavi, M.; Eagle, G.; Lane, R. Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients. Curr. Alzheimer Res. 2009, 6, 4–14. [Google Scholar] [CrossRef]
- Ferris, S.; Nordberg, A.; Soininen, H.; Darreh-Shori, T.; Lane, R. Progression from mild cognitive impairment to Alzheimer’s disease: Effects of gender, butyrylcholinesterase genotype and rivastigmine treatment. Pharmacogenet Genomics. 2009, 19, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Venneri, A.; Lane, R. Effects of cholinesterase inhibition on brain white matter volume in Alzheimer’s disease. NeuroReport 2009, 2, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.-L.; Kao, Y.-H.; Chou, M.-C.; Yang, Y.-H. Cerebral white matter changes on therapeutic response to rivastigmine in Alzheimer’s disease. J. Alzheimer Dis. 2016, 54, 351–357. [Google Scholar] [CrossRef]
- Fields, R.D.; Dutta, D.J.; Belgrad, J.; Robnett, J. Cholinergic signaling in myelination. Glia 2017, 65, 687–698. [Google Scholar] [CrossRef]
- Darvish, S. Butyrylcholinesterase as diagnostic and therapeutic target in Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1173–1177. [Google Scholar] [CrossRef]
- Dubois, B.; Chupin, M.; Hampel, H.; Lista, S.; Cavedo, E.; Croisile, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1041–1049. [Google Scholar] [CrossRef]
- Cavedo, E.; Dubois, B.; Colliot, O.; Lista, S.; Croisile, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; Rouaud, O.; et al. Reduced regional cortical thickness rate of change in donepezil-treated subjects with suspected prodromal Alzheimer’s disease. J. Clin. Psychiatry 2016, 77, e1631–e1638. [Google Scholar] [CrossRef]
- Cavedo, E.; Grothe, M.J.; Colliot, O.; Lista, S.; Chupin, M.; Dormont, D.; Houot, M.; Lehéricy, S.; Teipel, S.; Dubois, B.; et al. Reduced basal forebrain atrophy progression in a randomized donepezil trial in prodromal Alzheimer’s disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.R.; Charles, H.C.; Doraiswamy, P.M.; Mintzer, J.; Weisler, R.; Yu, X.; Perdomo, C.; Ieni, J.R.; Rogers, S. Randomized, placebo-controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer’s disease. Am. J. Psychiatry 2003, 160, 2003–2011. [Google Scholar] [CrossRef]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Bruno, M.A.; Allard, S.; Leon, W.; Iulita, M.F. Cholinergic involvement in alzheimer’s disease. A link with NGF maturation and degradation. J. Mol. Neurosci. 2010, 40, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mobley, W.C. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: Converging insights from alternative hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Latina, V.; Caioli, S.; Zona, C.; Ciotti, M.T.; Amadoro, G.; Calissano, P. Impaired NGF/TrkA Signaling Causes Early AD-Linked Presynaptic Dysfunction in Cholinergic Primary Neurons. Front. Cell. Neurosci. 2017, 11, 1–23. [Google Scholar] [CrossRef]
- Counts, S.E.; Mufson, E.J. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J. Neuropathol. Exp. Neurol. 2005, 64, 263–272. [Google Scholar] [CrossRef]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase inhibitor therapy in Alzheimer’s disease: The limits and tolerability of irreversible CNS-selective acetylcholinesterase inhibition in primates. J. Alzheimer’s Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef]
- Deutsch, J.A. The Cholinergic Synapse and the Site of Memory. Science 1971, 174, 788–794. [Google Scholar] [CrossRef]
- Janeczek, M.; Gefen, T.; Samimi, M.; Kim, G.; Weintraub, S.; Bigio, E.; Rogalski, E.; Mesulam, M.-M.; Geula, C. Variations in acetylcholinesterase activity within human cortical pyramidal neurons across age and cognitive trajectories. Cereb. Cortex 2018, 28, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L.; Pontecorvo, M.J.; Flicker, C. The cholinergic hypothesis: A historical overview, current perspective, and future irections. Ann. N. Y. Acad. Sci. 1985, 444, 332–358. [Google Scholar] [CrossRef] [PubMed]
- Drachmann, D.A.; Leavitt, J. Human memory and the cholinergic system. A relationship to aging? Arch. Neurol. 1974, 30, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Rogers, G.; Peters, J.; Anderson, R.; Hoyle, M.; Miners, A.; Moxham, T.; Davis, S.; Thokala, P.; Wailoo, A.; et al. The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of technology appraisal no. 111): A systematic review and Economic model. Health Technol. Assess. 2012, 16, 1–470. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.H.; Pirttila, T.; Dartigues, J.F.; Everitt, B.; van Baelen, B.; Schwalen, S.; Kavanagh, S. Treatment with galantamine and time to nursing home placement in Alzheimer’s disease patients with and without cerebrovascular disease. Int. J. Geriatr. Psychiatry 2009, 24, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Hommet, C.; Novella, J.; Auriacombe, S.; Vercelletto, M.; Berrut, G.; Belliard, S.; Desmidt, T.; Ceccaldi, M.; Centre, C.; Tours, C. Les traitements symptomatiques à partir des Centres mémoire ressources. Geriatr. Psychol. Neuropsychiatr. du Vieil. 2016, 14, 274–286. [Google Scholar]
- Krolak-salmon, P.; Dubois, B.; Vandel, P. France will no more reimburse available symptomatic drugs against Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 66, 425–427. [Google Scholar] [CrossRef]
- Loveman, E.; Green, C.; Kirby, J.; Takeda, A.; Picot, J.; Payne, E.; Clegg, A. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol. Assess. 2006, 10, 1–160. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opin Investig Drugs 2016, 25, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.J.; Koeppe, R.A.; Meltzer, C.C.; Constantine, G.; Davis, J.G.; Mathis, C.A.; et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzeimer’s disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, D.E.; Minoshima, S.; Frey, K.A.; Foster, N.L.; Kilbourn, M.R.; Koeppe, R.A. Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann. Neurol. 2000, 48, 391–395. [Google Scholar] [CrossRef]
- Ota, T.; Shinotoh, H.; Fukushi, K.; Kikuchi, T.; Sato, K.; Tanaka, N.; Shimada, H.; Hirano, S.; Miyoshi, M.; Arai, H.; et al. Estimation of plasma IC50 of donepezil for cerebral acetylcholinesterase inhibition in patients with Alzheimer disease using positron emission tomography. Clin. Neuropharmacol. 2010, 33, 74–78. [Google Scholar] [CrossRef]
- Kaasinen, V.; Någren, K.; Järvenpää, T.; Roivainen, A.; Yu, M.; Oikonen, V.; Kurki, T.; Rinne, J.O. Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J. Clin. Psychopharmacol. 2002, 22, 615–620. [Google Scholar] [CrossRef]
- Kadir, A.; Darreh-Shori, T.; Almkvist, O.; Wall, A.; Grut, M.; Strandberg, B.; Ringheim, A.; Eriksson, B.; Blomquist, G.; Långström, B.; et al. PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiol. Aging 2008, 29, 1204–1217. [Google Scholar] [CrossRef]
- Imbimbo, B.P. Pharmacodynamic-tolerability relationships of cholinesterase inhibitors for Alzheimer’s disease. CNS Drugs 2001, 15, 375–390. [Google Scholar] [CrossRef]
- Jann, M.W.; Shirley, K.L.; Small, G.W. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739. [Google Scholar] [CrossRef]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of alzheimer’s disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Sugimoto, H.; Iimura, Y.; Yamanishi, Y.; Yamatsu, K. Synthesis and structure-activity relationships of acetylcholinesterase inhibitors: 1-Benzyl-4-[(5,6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine hydrochloride and related Compounds. J. Med. Chem. 1995, 38, 4821–4829. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, T.; Kewitz, H. Selective inhibition of human acetylcholinesterase by galanthamine in vitro and in vivo. Life Sci. 1990, 46, 1553–1558. [Google Scholar] [CrossRef]
- Mannens, G.S.; Snel, C.A.; Hendrickx, J.; Verhaeghe, T.; le Jeune, L.; Bode, W.; van Beijsterveldt, L.; Lavrijsen, K.; Leempoels, J.; van Osselaer, N.; et al. The metabolism and excretion of galantamine in rats, dogs, and humans. Drug Metab. Dispos. 2002, 30, 553–563. [Google Scholar] [CrossRef]
- Plaitakis, A.; Duvoisin, R. Homer’s moly identified as Galanthus nivalis L.: Physiologic antidote to stramonium poisoning. Clin. Neuropharmacol. 1983, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Samochocki, M.; Zerlin, M.; Jostock, R.; Groot Kormelink, P.J.; Luyten WH, M.L.; Albuquerque, E.X.; Maelicke, A. Galantamine is an allosterically potentiating ligand of the human alpha4/beta2 nAChR. Acta Neurol Scand Suppl. 2000, 176, 68–73. [Google Scholar] [CrossRef]
- Schrattenholz, A.; Pereira, E.F.; Roth, U.; Weber, K.-H.; Albuquerque, E.X.; Maelicke, A. Agonist responses of neuronal nicotinic acetylcholine receptors are potentiated by a novel class of allosterically acting ligands. Mol. Pharmacol. 1996, 49, 1–6. [Google Scholar]
- Lilienfeld, S. Galantamine—A novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. 2002, 8, 159–176. [Google Scholar] [CrossRef]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef]
- Enz, A.; Floersheim, P. Cholinesterase inhibitors: An overview of their mechanisms of action. In Alzheimer’s Disease. Therapeutic Strategies; Giacobini, E., Becker, R., Eds.; Birkhauser: Boston, MA, USA, 1994; pp. 211–215. [Google Scholar]
- Lane, R.; Darreh-Shori, T. Understanding the benefits and detrimental effects of donepezil and rivastigmine to improve their therapeutic value. J. Alzheimer’s Dis. 2015, 44, 1039–1062. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2005, 9, 101–124. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim Care Companion CNS Disord 2013, 15, PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Nielsen, J.A.; Villalobos, A.; Chapin, D.; Jones, S.B.; Hubbard, S.T.; Shalaby, I.A.; Ramirez, A.; Nason, D.; White, W.F. Pharmacology of selective acetylcholinesterase inhibitors: Implications for use in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 486, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.N.; Brimijoin, S. Cholinesterases and the fine line between poison and remedy. Biochem. Pharmacol. 2018, 153, 205–216. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef]
- Becker, R.E.; Colliver, J.A.; Markwell, S.J.; Moriearty, P.L.; Unni, L.K.; Vicari, S. Double-Blind, Placebo-Controlled Study of Metrifonate, an Acetylcholinesterase Inhibitor, for Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 1996, 10, 124–131. [Google Scholar] [CrossRef]
- López-Arrieta, J.M.; Schneider, L. Metrifonate for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, CD003155. [Google Scholar] [CrossRef]
- Jewsbury, J.M.; Cooke, M.J.; Weber, M.C. Field trial of metrifonate in the treatment and prevention of schistosomiasis infection in man. Ann. Trop. Med. Parasitol. 1977, 71, 67–83. [Google Scholar] [CrossRef]
- Nordgren, I.; Bergström, M.; Holmstedt, B.; Sandoz, M. Transformation and action of metrifonate. Arch. Toxicol. 1978, 41, 31–41. [Google Scholar] [CrossRef]
- Holmstedt, B.; Nordgren, I.; Sandoz, M.; Sundwall, A. Metrifonate. Arch. Toxicol. 1978, 41, 3–29. [Google Scholar] [CrossRef]
- Metcalf, R.; Fukuto, R.; March, R. Toxic action of Dipterex and DDVP to the house fly. J. Econ. Entomol. 1959, 52, 44–49. [Google Scholar] [CrossRef]
- Pacheco, G.; Palacios-Esquivel, R.; Moss, D.E. Cholinesterase inhibitors proposed for treating dementia in Alzheimer’s disease: Selectivity toward human brain acetylcholinesterase compared with butyrylcholinesterase. J. Pharmacol. Exp. Ther. 1995, 74, 767–770. [Google Scholar]
- Kobayashi, H.; Nakano, T.; Moss, D.E.; Suzuki, T. Effects of a central anticholinesterase, methanesulfonyl fluoride on the cerebral cholinergic system and behavior in mice: Comparison with an organophosphate DDVP. J. Heal. Sci. 1999, 45, 191–202. [Google Scholar] [CrossRef]
- Unni, L.; Womack, C.; Hannant, M.; Becker, R. Pharmacokinetics and pharmacodynamics of metrifonate in humans. Methods Find. Exp. Clin. Pharmacol. 1994, 16, 285–289. [Google Scholar]
- Hallak, M.; Giacobini, E. A comparison of the effects of two inhibitors on brain cholinesterase. Neuropharmacology 1987, 26, 521–530. [Google Scholar] [CrossRef]
- Zalewska, Z.; Rakowska, I.; Matraszek, G.; Sitkiewicz, D. Effect of dichlorvos on some enzymes activites of the rat brain during postnatal development. Neuropatol. Pol. 1977, 15, 255–262. [Google Scholar]
- Caroldi, S.; Lotti, M. Delayed neurotoxicity caused by a single masssive dose of dichlorvos to adult hens. Toxicol. Lett. 1981, 9, 157–159. [Google Scholar] [CrossRef]
- Vasilescu, C.; Florescu, A. Clinical and electrophysiological study of neuropathy after organophosphorus compounds poisoning. Arch. Toxicol. 1980, 43, 305–315. [Google Scholar] [CrossRef]
- Desi, I.; Nagymajtenyi, L. Neurotoxicologic investigations of the pesticide dichlorvos (DDVP): Effects on the central and peripheral nervous system. Toxicology 1988, 49, 141–148. [Google Scholar] [CrossRef]
- Sitkiewicz, D.; Zalewska, Z. Aktywność oksydazy cytochromowej i dehydrogenazy bursztynianowej mózgu szczura po zatruciu fosforoorganicznymi insektycydami dichlorfosem i trichlorfonem [The activity of cytochrome oxidase and succinate dehydrogenase in rat brain mitochondria following trichlorphon and dichlorvos intoxication]. Neuropathol. Pol. 1975, 13, 279–280. [Google Scholar]
- Lotti, M. Promotion of organophosphate induced delayed polyneuropathy by certain esterase inhibitors. Toxicology 2002, 181–182, 245–248. [Google Scholar] [CrossRef]
- Dou, K.-X.; Tan, M.-S.; Tan, C.-C.; Cao, X.-P.; Hou, X.-H.; Guo, Z.-H.; Tan, L.; Mok, V.; Yu, J.-T. Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: A network meta-analysis of 41 controlled trials. Alzheimer’s Res. Thera. 2018, 10, 126. [Google Scholar] [CrossRef]
- Chase, T.N.; Farlow, M.R.; Clarence-Smith, K. Donepezil plus solifenacin (CPC-201) treatment of Alzheimer’s disease. Neurotherapeutics 2017, 14, 405–416. [Google Scholar] [CrossRef]
- Karami, A.; Eriksdotter, M.; Kadir, A.; Almkvist, O.; Nordberg, A.; Darreh-Shori, T. CSF cholinergic index, a new biomeasure of treatment effect in patients with Alzheimer’s disease. Front. Mol. Neurosci. 2019, 12, 239. [Google Scholar] [CrossRef]
- Moss, D.E.; Berlanga, P.; Hagan, M.M.; Sandoval, H.; Ishida, C. Methanesulfonyl fluoride (MSF): A double-blind, placebo-controlled study of safety and efficacy in the treatment of senile dementia of the Alzheimer type. Alzheimer Dis. Assoc. Disord. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- Moss, D.E.; Rodriguez, L.; Selim, S.; Ellett, S.; Devine, J.; Steger, R. The sulfonyl fluorides: CNS selective cholinesterase inhibitors with potential value in Alzheimer’s disease? In Neurology and Neurobiology 18: Senile Dementia of the Alzheimer Type; Hutton, J.T., Kenny, A.D., Eds.; Alan, R. Liss: New York, NY, USA, 1985; pp. 337–350. [Google Scholar]
- Moss, D.E.; Fariello, R.G.; Sahlmann, J.; Sumaya, I.; Pericle, F.; Braglia, E. A randomized phase I study of methanesulfonyl fluoride, an irreversible cholinesterase inhibitor, for the treatment of Alzheimer’s disease. Br. J. Clin. Pharmacol. 2013, 75, 1231–1239. [Google Scholar] [CrossRef]
- Malin, D.H.; Plotner, R.E.; Radulescu, S.J.; Ferebee, R.N.; Lake, J.R.; Negrete, P.G.; Schaefer, P.J.; Crothers, M.K.; Moss, D.E. Chronic methanesulfonyl fluoride enhances one-trial per day reward learning in aged rats. Neurobiol Aging. 1993, 14, 393–395. [Google Scholar] [CrossRef]
- Myers, D.; Kemp, A. Inhibition of esterases by the fluorides of organic acids. Nature 1954, 173, 33–34. [Google Scholar] [CrossRef]
- Kitz, R.; Wilson, I. Esters of methanesulfonic acids as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar]
- Fahrney, D.; Gold, A. Sulfonyl fluorides as inhibitors of esterases. J. Am. Chem. Soc. 1963, 85, 997–1000. [Google Scholar] [CrossRef]
- Snow, A.W.; Barger, W.R. A chemical comparison of methanesulfonyl fluoride with organofluorophosphorus ester anticholinesterase compounds. Chem. Res. Toxicol. 1988, 1, 379–384. [Google Scholar] [CrossRef]
- Osman, K. Sulfonyl Fluorides and the Promotion of Diisopropyl Fluorophosphate Neuropathy. Fundam. Appl. Toxicol. 1996, 33, 294–297. [Google Scholar] [CrossRef]
- Moss, D.; Keathley, S. Pilot Study to Test Sulfnates’ Ability to Provide Prophylaxis Against Nerve Agents; Technical Report; (Contract No. DAMD 17-87-C-7064); The U.S. Army Medical Research and Development Command: Frederick, MD, USA, 1 July 1988. [Google Scholar]
- Moss, D.; Rodriguez, L.; Herndon, W.; Vincenti, S.; Camarena, M. Sulfonyl fluorides as possible therapeutic agents in Alzheimer’s disease: Structure/activity relationships as CNS selective cholinesterase inhibitors. In Alzheimer’s and Parkinson’s Disease: Strategies in Research and Development; Fisher, A., Lachman, C., Hanin, I., Eds.; Plenum Press: New York, NY, USA, 1986; pp. 551–556. [Google Scholar]
- Moss, D.; Kobayashi, H.; Pacheco, G.; Palacios, R.; Perez, R. Methanesulfonyl fluoride: A CNS selective cholinesterase inhibitor. In Current Research in Alzheimer Therapy: Cholinesterase Inhibitors; Giacobini, E., Becker, R., Eds.; Taylor and Francis: New York, NY, USA, 1988; pp. 305–314. [Google Scholar]
- Borlongan, C.V.; Sumaya, I.C.; Moss, D.E. Methanesulfonyl fluoride, an acetylcholinesterase inhibitor, attenuates simple learning and memory deficits in ischemic rats. Brain Res. 2005, 1038, 50–58. [Google Scholar] [CrossRef]
This entry is adapted from the peer-reviewed paper 10.3390/ijms21103438