 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sumiko Mochida | + 2602 word(s) | 2602 | 2021-04-22 08:49:25 | | | |
| 2 | Vivi Li | Meta information modification | 2602 | 2021-04-23 03:34:30 | | |
Video Upload Options
Presynaptic Ca2+ entry occurs through voltage-gated Ca2+ (CaV) channels which are activated by membrane depolarization. Depolarization accompanies neuronal firing and elevation of Ca2+ triggers neurotransmitter release from synaptic vesicles. For synchronization of efficient neurotransmitter release, synaptic vesicles are targeted by presynaptic Ca2+ channels forming a large signaling complex in the active zone. The presynaptic CaV2 channel gene family (comprising CaV2.1, CaV2.2, and CaV2.3 isoforms) encode the pore-forming α1 subunit. The cytoplasmic regions are responsible for channel modulation by interacting with regulatory proteins.
1. Introduction
Presynaptic Ca2+ entry into the active zone (AZ) occurs through voltage-gated Ca2+ (CaV) channels which are activated membrane depolarization and triggers synchronous neurotransmitter release from synaptic vesicles (SVs). Multiple mechanisms regulate the function of presynaptic Ca2+ channels [1][2][3][4]. The channel activity for opening, closing, or inactivation in response to membrane depolarization changes every few milliseconds during and after neuronal firing, resulting in control of synaptic strength [3][4]. Following a brief overview of Ca2+ channel structure/function, this article reviews the molecular and cellular mechanisms that modulate the activity of presynaptic Ca2+ channels in the regulation of neurotransmitter release and in the induction of short-term synaptic plasticity. To understand the physiological role of Ca2+ channel modulation in the regulation of synaptic transmission, a model synapse formed between sympathetic, superior cervical ganglion (SCG) neurons in culture was employed for functional study of channel interaction with G proteins, SNARE proteins, and Ca2+-binding proteins which sense residual Ca2+ in the AZ after the arrival of an action potential (AP).
2. Presynaptic Ca2+ Channels
Ca2+ currents have diverse physiological roles and different pharmacological properties. Early investigations revealed distinct classes of Ca2+ currents which were identified with an alphabetical nomenclature [5]. P/Q-type, N-type, and R-type Ca2+ currents are observed primarily in neurons, require strong depolarization for activation [6], and are blocked by specific polypeptide toxins from snail and spider venoms [7]. P/Q-type and N-type Ca2+ currents initiate neurotransmitter release at most fast synapses [1][8][9]. The Ca2+ channels are composed of four or five distinct subunits (Figure 1a) [8][10]. The α1 subunit incorporates the conduction pore, the voltage sensors and gating apparatus, and target sites of toxins and intracellular regulators. The α1 subunit is composed of about 2000 amino acid residues and is organized in four homologous domains (I–IV) (Figure 1b). Each domain consists of six transmembrane α helices (S1 through S6) and a membrane-associated P loop between S5 and S6. The S1 through S4 segments serve as the voltage sensor module, whereas transmembrane segments S5 and S6 in each domain and the P loop between them form the pore module [11]. The intracellular segments serve as a signaling platform for Ca2+-dependent regulation of neurotransmission, as discussed below.
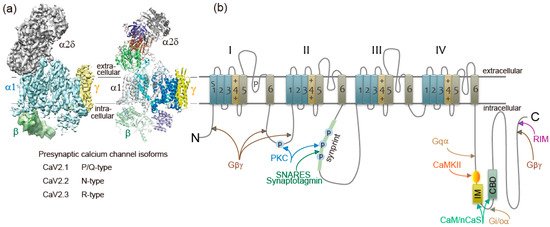
Figure 1. Ca2+ channel structure and organization. (a) The subunit composition and structure of high-voltage-activated Ca2+ channels. The cryo-EM structure of the rabbit voltage-gated Ca2+ channel Cav1.1 complex at a nominal resolution of 4.2 Å. The overall EM density map on the left is colored according to different subunits. The structure model on the right is color-coded for distinct subunits. Reproduced from [12]. (b) The α1 subunit consists of four homologous domains (I-IV), each consisting of six transmembrane segments (S1-S6). S1–S4 represents the voltage-sensing module. S5–S6 represents the pore-forming unit. The large intracellular loops linking the different domains of the α1 subunit serve as sites of interaction of different regulatory proteins important for channel regulation, including G-protein (Gβγ, Gα), RIM, SNARE proteins, and synaptotagmin at the synprint site (shown in green bar), calmodulin (CaM), and neuronal Ca2+ sensor proteins (nCaS) at the IQ-like motif, which begins with the sequence isoleucine-methionine (IM) instead of isoleucine-glutamine (IQ) and the nearby downstream CaM-binding domain (CBD), calmodulin kinase II (CaMKII), and protein kinase C (PKC). Adapted from [4].
Ca2+ channel α1 subunits are encoded by ten distinct genes in mammals, which are divided into three subfamilies by sequence similarity [2][8][13]. The CaV2 subfamily members CaV2.1, CaV2.2, and CaV2.3 channels conduct P/Q-type, N-type, and R-type Ca2+ currents, respectively [2][8][9][13].
CaV channels are complexes of a pore-forming α1 subunit and auxiliary subunits. Skeletal muscle CaV channels have three distinct auxiliary protein subunits [8] (Figure 1a), the intracellular β subunit, the disulfide-linked α2δ subunit complex, and the γ subunit having four transmembrane segments. In contrast, brain neuron CaV2 channels are composed of the pore-forming α1 and the auxiliary β subunit [14]. The auxiliary subunits of Ca2+ channels have an important influence on their function [15][16]. The CaVβ subunit shifts their kinetics and voltage dependence of activation and inactivation [15][16]. Cell surface expression of the α1 subunits is enhanced by the CaVβ subunit [15][16]. The α2δ subunits are potent modulators of synaptic transmission. The α2δ subunits increase not only Cav1.2 but also Cav2.2, Cav2.1 currents, suggesting that the α2δ subunits enhance trafficking of the CaV channel complex [17]. Expression of α2δ subunits also appears to play a role in setting release probability [18]. Further details of these regulatory interactions are discussed below.
3. Intracellular Molecules Modulate Presynaptic Ca2+ Channels Activity
3.1. G Proteins
Presynaptic Ca2+ currents are reduced in magnitude by activation of G protein-coupled receptors for neurotransmitters at nerve terminals [19][20]. Gβγ subunits released from heterotrimeric G proteins of the Gi/Go class [19][20] bind directly to α1 subunits of the N-type Ca2+ channel [21][22] at the N terminus [23], the intracellular loop connecting domains I and II [21][24], and at the C terminus [25] (Figure 1b). Gβγ causes a positive shift in the voltage dependence of activation of the Ca2+ current [26][27][28]. The Gβγ-induced reduction of Ca2+ currents can be reversed by strong positive depolarization [26][27][28]. Reversal of this inhibition by depolarization provides a point of intersection between chemical and electrical signal transduction at the synapse and can potentially provide novel forms of short-term synaptic plasticity that do not rely on residual Ca2+.
The subtype of CaVβ can influence the extent and kinetics of Gβγ mediated inhibition and this regulation also depends on the subtype of Gβ involved [29][30]. Gβγ interacts with multiple sites on the N-terminus, I–II linker, and the C-terminus of the α1 subunit. Binding of Gβγ causes a conformational shift that promotes interaction of the N-terminus “inhibitory module” with the initial one-third of the I–II-linker. Strong membrane depolarization leads to unbinding of Gβγ and loss of interaction between the N-terminus and the I–II linker. This depends upon binding of CaVβ subunit to the α interaction domain (AID) on the I–II linker. In the absence of CaVβ1 subunit binding with tryptophan mutation in the AID (W391) of the CaV2.2 α1 subunit, Ca2+ channel inhibition still occurs but cannot be reversed by strong depolarization. CaVβ2a, that is palmitoylated at two N-terminal cysteine residues, can still bind to the α1 subunit and permit voltage-dependent relief of the inhibition [31]. It is possible that binding of CaVβ1 to the AID induces a rigid α-helical link with domain IS6, and this transmits the movement of the voltage-sensor and activation gate to the I–II linker to alter the Gβγ binding pocket at depolarized potentials [32].
Specific Gβ subunits have been shown to be responsible for the CaV2 channel modulation in different neurons. In rat SCG neurons CaV2.2 channels are differentially modulated by different types of Gβ subunits, with Gβ1 and Gβ2 being most effective, Gβ5 showing weaker modulation, and Gβ3 and Gβ4 being ineffective [33][34][35]. In contrast, in rat stellate ganglion neurons, Gβ2 and Gβ4 but not Gβ1 subunit are responsible for the coupling of CaV2.2 channels with noradrenaline receptors [36]. In the transfected human embryonic kidney tsA-201 cell line, CaV2.2 channel inhibition, with Gβ1 and Gβ3 being more effective than Gβ4 and Gβ2, and no significant modulation being induced by Gβ5 [37]. Gβ subunit-induced inhibition of CaV2.1 channel differed from those observed with the CaV2.2 channel. CaV2.1 channels exhibited more rapid rates of recovery from inhibition than those observed with CaV2.2 channels, on average, twice as rapidly for the CaV2.1 channels, indicating that Gβ binding to this channel subtype is less stable [37].
Regulation of the CaV2.2 channels also involves the interplay between Ca2+ channels and G protein interaction. Syntaxin-1A, a presynaptic plasma membrane protein, is required for G protein inhibition of presynaptic Ca2+ channels [38]. Physical interaction between syntaxin-1A and Ca2+ channels is a prerequisite for tonic Gβγ modulation of CaV2.2 channels, suggesting that syntaxin-1A mediates a colocalization of Gβγ subunits and CaV2.2 channels, thus resulting in a more effective G protein coupling to, and regulation of, the channel. The interactions between syntaxin, G proteins, and CaV2.2 channels are part of the structural specialization of the presynaptic terminal [39].
G proteins also induce voltage-independent inhibition of CaV2 channels through intracellular signaling pathways [1][19][40]. This often involves the Gq family of G proteins, which regulate the levels of phosphatidylinositide lipids by inducing hydrolysis of phosphatidylinositol bisphosphate via activation of phospholipase C enzymes [41]. Acetylcholine release from rat sympathetic neurons is reduced through this pathway via presynaptic muscarinic receptors activation [42].
3.2. Active Zone Proteins
Rab-interacting molecule (RIM), an AZ protein required for SVs docking and priming [43][44][45][46][47][48], and synaptic plasticity [49], interacts with the C-terminal cytoplasmic tails of CaV2.1 and CaV2.2 channels [46][48][50][51] (Figure 1b). The interaction is essential for recruiting Ca2+ channels to the presynaptic AZ [46] and determines channel density and SVs docking at the presynaptic AZ [48]. RIM-binding proteins, RIM-BPs, also interact with CaV2.1 and CaV2.2 channels [51], and are selectively required for high-fidelity coupling of AP-induced Ca2+ influx to Ca2+-stimulated SVs exocytosis [52]. The tripartite complex of RIM, RIM-BPs, and C-terminal tails of the CaV2 channels regulate the recruitment of CaV2 channels to AZs. Interaction of RIM with CaVβ subunits shifts the voltage dependence of inactivation to more positive membrane potentials, increasing Ca2+ channel activity [53]. In contrast, CaVβ subunits interaction with CAST/ERC2 shifts the voltage dependence of activation to more negative membrane potentials [54]. Positive regulation of presynaptic Ca2+ channel activity by RIM and CAST/ERC2, in addition to their function in SVs docking, increase the release probability of SVs docked close to CaV2 channels. Furthermore, Munc13, required for SVs priming, controls CaV2 channels shortly after AP firing to guarantee transmitter release for continuous neural activity [55].
3.3. t-SNAREs and Synaptotagmin-1
SV (v)-SNARE synaptobrevin 2 and presynaptic plasma membrane (t)-SNAREs syntaxin-1 and SNAP-25 are required for fusion of SVs with a plasma membrane to release neurotransmitters [56]. Both CaV2.1 and CaV2.2 channels at the presynaptic nerve terminals colocalize densely with syntaxin-1A [57][58][59], and also form a complex of with SNARE proteins [60][61][62] dependent on Ca2+ with maximal binding at 20 μM and reduced binding at lower or higher concentrations of Ca2+ [63]. The t-SNARE proteins syntaxin-1A and SNAP-25, but not the v-SNARE synaptobrevin, bind to the intracellular loop between domains II and III of the α1 subunit of CaV2.2 (amino acid residues 718-963) named as the synprint site (Figure 1b) [64][65]. CaV2.1 channels have an analogous synprint site, and different channel isoforms have distinct interactions with syntaxin and SNAP-25 [66][67], suggesting specialized regulatory properties for synaptic modulation.
t-SNAREs interacting with presynaptic CaV2.1 and CaV2.2 channels regulate channel activity (Figure 3a). Syntaxin-1A or SNAP-25 shifts the voltage dependence of inactivation toward more negative membrane potentials and reduces the availability of the channels to open [68][69][70]. Coexpression of SNAP-25 can reverse the inhibitory effects of syntaxin-1A [69][71]. The transmembrane region of syntaxin-1A and only a short segment within the H3 helix are critical for channel modulation [72], whereas the synprint site binds to the entire H3 helix in the cytoplasmic domain of syntaxin-1A [63][64][72]. Deletion of the synprint site weakened the modulation of the channels by syntaxin-1A, but did not abolish it, arguing that the synprint site acts as an anchor in facilitating channel modulation but is not required absolutely for modulatory action.
Dependent on Ca2+ concentration, syntaxin-1 interacts with either the synprint site or synaptotagmin-1; at low Ca2+ concentrations, syntaxin-1 binds synprint, while at higher concentrations (>30 μM) it associates with synaptotagmin-1 [63]. Synaptotagmin-1, -2, and -9 serve as the Ca2+ sensors for the fast, synchronous neurotransmitter release [56][73][74]. The Ca2+ binding site C2B domain of synaptotagmin-1 interacts with the synprint sites of both CaV2.1 and CaV2.2 channels (Figure 1b) [75]. Synaptotagmin-1 can relieve the inhibitory effects of SNAP-25 on CaV2.1 channels [70][76]. Relief of Ca2+ channel inhibition by the formation of the synaptotagmin/SNARE complex favors Ca2+ influx. This is a potential mechanism to increase the release probability of SVs docked close to CaV2 channels [4].
Interaction of syntaxin-1A and SNAP-25 with the synprint site is controlled by phosphorylation of the synprint site with protein kinase C (PKC) (Figure 1b) [65] and Ca2+/calmodulin-dependent protein kinase II (CaMKII) [77]. The negative shift of steady-state inactivation of CaV2.2 channels caused by syntaxin is blocked by PKC phosphorylation [65][71]. Thus, phosphorylation of the synprint site may serve as a biochemical switch controlling the SNARE-synprint interaction.
3.4. Ca2+-Sensor Proteins
Ca2+ elevation regulates CaV2.1 channels activity by its binding to CaM [8][78][79][80][81] and related neuron-specific Ca2+-binding proteins, CaBP1, VILIP-2 [82][83][84], and NCS-1 (frequenin) [85]. The presynaptic CaV2.1 channel proteins consist of a pore-forming α1 subunit associated with β, and possibly α2δ subunits (Figure 1a) [86]. The intracellular C terminus of the α1 subunit [81] called the IQ-like motif, which begins with the sequence isoleucine-methionine (IM) instead of isoleucine-glutamine (IQ), and the nearby downstream CaM-binding domain (CBD) are the interacting sites with these Ca2+-binding proteins (Figure 1b). Displacement with alanine in the IQ-like domain inhibited Ca2+-dependent CaV2.1 channels facilitation [78][81], whereas deletion of CBD inhibited Ca2+-dependent CaV2.1 channels inactivation [79][80][81][83][84]. Ca2+/CaM-dependent inactivation of CaV2.1 channels, dependent on global elevations of Ca2+, is observed in transfected cells overexpressing CaV2.1 channels [78][79][80] and in the nerve terminals of the calyx of Held [87][88] where CaV2.1 channels are densely localized. In contrast, the large neuronal cell bodies of Purkinje neurons [89] or SCG neurons [90] rarely show Ca2+-dependent CaV2.1 channels inactivation.
4. Synchronous Neurotransmitter Release Regulated by Ca2+ Channel/SNARE Proteins Complex
Synprint peptides derived from CaV2.2 channels reduced transmitter release from the microinjected presynaptic SCG neurons in culture, due to competitive uncoupling of the endogenous Ca2+ channel-SNARE proteins interaction in nerve terminals [91]. Synprint peptides selectively inhibited fast synchronous synaptic transmission, while they increased late asynchronous release (Figure 2b). Similarly, synprint peptides reduced transmitter release from embryonic Xenopus spinal neurons [92]. Increasing the external Ca2+ concentration effectively rescued this inhibition, implying that synprint peptides competitively displaces docked SVs away from Ca2+ channels, and this effect can be overcome by increasing Ca2+ influx into presynaptic terminals [92].
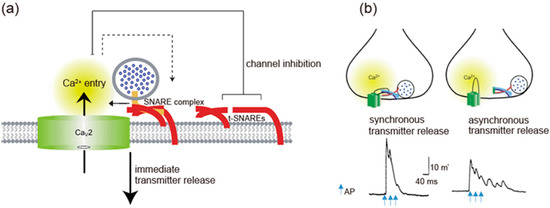
Figure 2. Spatial regulation of transmitter release by the I-II loop interaction with SNAREs. (a) The I-II loop interacts with t-SNAREs, resulting in inhibition of Ca2.2 channels opening. Once AP opens the channels, an increase in Ca2+ mediates interaction with SNAREs complex and induces transmitter release. Adapted from [4]. (b) Triple APs induces a large synchronous transmitter release from the first AP. In contrast, asynchronous transmitter release was observed in the presence of 130 μM synprint peptide (see Figure 1b). Adapted from [91].
At the calyx of Held, presynaptic neurons express P/Q-, N- and R-type Ca2+ currents in postnatal day 7 rats. P/Q-type Ca2+ currents are more effective than N-type Ca2+ currents and R-type Ca2+ currents in eliciting neurotransmitter release [93][94][95]. The high efficiency of P/Q-type Ca2+ currents to initiate neurotransmitter release is correlated with the close localization of CaV2.1 channels near docked SVs [96], as shown by immunocytochemistry [97], suggesting localization of CaV2 channels determines the efficiency of neurotransmitter release in response to neural activity.
CaV2 channels interaction with SNARE proteins, that is dependent on Ca2+ concentration [63], have two opposing effects: at the pre-firing state synaptic transmission is blocked by enhancing CaV2 channels inactivation, whereas immediately after AP firing tethering SVs near the point of Ca2+ entry enhances synaptic transmission. The overexpression of a syntaxin mutant that is unable to regulate CaV2.2 channels, but still binds to them [72], increased the efficiency of synaptic transmission at Xenopus neuromuscular junctions, as reflected in increased quantal content [98]. In contrast, injected synprint peptides reduced the basal efficiency of synaptic transmission, as reflected in reduced quantal content of synaptic transmission [98]. These results demonstrate a bidirectional regulation of synaptic transmission in vivo by interactions of CaV2.2 channels with SNARE proteins.
References
- Dunlap, K.; Luebke, J.I.; Turner, T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995, 18, 89–98.
- Snutch, T.P.; Reiner, P.B. Ca2+ channels: Diversity of form and function. Curr. Opin. Neurobiol. 1992, 2, 247–253.
- Tedford, H.W.; Zamponi, G.W. Direct G protein modulation of Cav2 calcium channels. Pharm. Rev. 2006, 58, 837–862.
- Catterall, W.A.; Few, A.P. Calcium channel regulation and presynaptic plasticity. Neuron 2008, 59, 882–901.
- Tsien, R.W.; Lipscombe, D.; Madison, D.V.; Bley, K.R.; Fox, A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988, 11, 431–438.
- Tsien, R.W.; Ellinor, P.T.; Horne, W.A. Molecular diversity of voltage-dependent Ca2+ channels. Trends Pharm. Sci. 1991, 12, 349–354.
- Miljanich, G.P.; Ramachandran, J. Antagonists of neuronal calcium channels: Structure, function, and therapeutic implications. Annu. Rev. Pharm. Toxicol. 1995, 35, 707–734.
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555.
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M. Calcium channel diversity and neurotransmitter release: The ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867.
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482.
- Frank, H.Y.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395.
- Wu, J.; Yan, Z.; Li, Z.; Yan, C.; Lu, S.; Dong, M.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 complex. Science 2015, 350, aad2395.
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535.
- Müller, C.S.; Haupt, A.; Bildl, W.; Schindler, J.; Knaus, H.G.; Meissner, M.; Meissner, B.; Striessnig, J.; Flockerzi, V.; Fakler, B.; et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14950–14957.
- Dolphin, A.C. Beta subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 2003, 35, 599–620.
- Hofmann, F.; Lacinova, L.; Klugbauer, N. Voltage-dependent calcium channels: From structure to function. Rev. Physiol. Biochem. Pharm. 1999, 139, 33–87.
- Davies, A.; Hendrich, J.; Van Minh, A.T.; Wratten, J.; Douglas, L.; Dolphin, A.C. Functional biology of the α2δ subunits of voltage-gated calcium channels. Trends Pharm. Sci. 2007, 28, 220–228.
- Hoppa, M.B.; Lana, B.; Margas, W.; Dolphin, A.C.; Ryan, T.A. α2δ a expression sets presynaptic calcium channel abundance and release probability. Nature 2012, 486, 122–125.
- Hille, B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994, 17, 531–536.
- Ikeda, S.R.; Dunlap, K. Voltage-dependent modulation of N-type calcium channels: Role of G protein subunits. Adv. Second Messenger Phosphoprot. Res. 1999, 33, 131–151.
- Hertlitze, S.; Garcia, D.E.; Mackie, K.; Hille, B.; Scheuer, T.; Catterall, W.A. Modulation of Ca2+ channels by G-protein βγ subunits. Nature 1996, 380, 258–262.
- Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 1996, 380, 255–258.
- Cantı, C.; Page, K.M.; Stephens, G.J.; Dolphin, A.C. Identification of residues in the N terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβγ. J. Neurosci. 1999, 19, 6855–6864.
- Zamponi, G.W.; Bourinet, E.; Nelson, D.; Nargeot, J.; Snutch, T.P. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature 1997, 385, 442–446.
- Li, B.; Zhong, H.; Scheuer, T.; Catterall, W.A. Functional role of a C-terminal G βγ-binding domain of Cav2.2 channels. Mol. Pharm. 2004, 66, 761–769.
- Bean, B.P. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 1989, 340, 153–156.
- Marchetti, C.; Carbone, E.; Lux, H.D. Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflug. Arch. 1986, 406, 104–111.
- Tsunoo, A.; Yoshii, M.; Narahashi, T. Block of calcium channels by enkephalin and somatostatin in neuroblastoma-glioma hybrid NG108-15 cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9832–9836.
- Canti, C.; Bogdanov, Y.; Dolphin, A.C. Interaction between G proteins and accessory subunits in the regulation of 1B calcium channels in Xenopus oocytes. J. Physiol. 2000, 527 Pt. 3, 419–432.
- Feng, Z.P.; Arnot, M.I.; Doering, C.J.; Zamponi, G.W. Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gβ isoforms. J. Biol. Chem. 2001, 276, 45051–45058.
- Dresviannikov, A.V.; Page, K.M.; Leroy, J.; Pratt, W.S.; Dolphin, A.C. Determinants of the voltage dependence of G protein modulation within calcium channel beta subunits. Pflug. Arch. 2009, 457, 743–756.
- Zamponi, G.W.; Currie, K.P. Regulation of CaV2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 2013, 1828, 1629–1643.
- García, D.E.; Li, B.; García-Ferreiro, R.E.; Hernández-Ochoa, E.O.; Yan, K.; Gautam, N.; Catterall, W.A.; Mackie, K.; Hille, B. G-protein beta-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J. Neurosci. 1998, 18, 9163–9170.
- Reyes-Vaca, A.; de la Cruz, L.; Garduño, J.; Arenas, I.; Garcia, D.E. Fast Inactivation of CaV2.2 Channels Is Prevented by the Gβ1 Subunit in Rat Sympathetic Neurons. J. Mol. Neurosci 2017, 63, 377–384.
- Hernández-Castellanos, J.M.; Vivas, O.; Garduño, J.; De la Cruz, L.; Arenas, I.; Elías-Viñas, D.; Mackie, K.; García, D.E. Gβ2 mimics activation kinetic slowing of CaV2.2 channels by noradrenaline in rat sympathetic neurons. Biochem. Biophys. Res. Commun. 2014, 445, 250–254.
- Mahmoud, S.; Yun, J.K.; Ruiz-Velasco, V. Gβ2 and Gβ4 participate in the opioid and adrenergic receptor-mediated Ca2+ channel modulation in rat sympathetic neurons. J. Physiol. 2012, 590, 4673–4689.
- Arnot, M.I.; Stotz, S.C.; Jarvis, S.E.; Zamponi, G.W. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J. Physiol. 2000, 527 Pt. 2, 203–212.
- Stanley, E.F.; Mirotznik, R.R. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature 1997, 385, 340–343.
- Jarvis, S.E.; Magga, J.M.; Beedle, A.M.; Braun, J.E.; Zamponi, G.W. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gβγ. J. Biol. Chem. 2000, 275, 6388–6394.
- Strock, J.; Diverse-Pierluissi, M.A. Ca2+ channels as integrators of G protein-mediated signaling in neurons. Mol. Pharm. 2004, 66, 1071–1076.
- Delmas, P.; Coste, B.; Gamper, N.; Shapiro, M.S. Phosphoinositide lipid second messengers: New paradigms for calcium channel modulation. Neuron 2005, 47, 179–182.
- Kubista, H.; Kosenburger, K.; Mahlknecht, P.; Drobny, H.; Boehm, S. Inhibition of transmitter release from rat sympathetic neurons via presynaptic M1 muscarinic acetylcholine receptors. Br. J. Pharm 2009, 156, 1342–1352.
- Koushika, S.P.; Richmond, J.E.; Hadwiger, G.; Weimer, R.M.; Jorgensen, E.M.; Nonet, M.L. A post-docking role for active zone protein Rim. Nat. Neurosci. 2001, 4, 997–1005.
- Schoch, S.; Mittelstaedt, T.; Kaeser, P.S.; Padgett, D.; Feldmann, N.; Chevaleyre, V.; Castillo, P.E.; Hammer, R.E.; Han, W.; Schmitz, F.; et al. Redundant functions of RIM1α and RIM2α in Ca2+-triggered neurotransmitter release. EMBO J. 2006, 25, 5852–5863.
- Gracheva, E.O.; Hadwiger, G.; Nonet, M.L.; Richmond, J.E. Direct interactions between C. elegans RAB-3 and Rim provide a mechanism to target vesicles to the presynaptic density. Neurosci. Lett. 2008, 444, 137–142.
- Kaeser, P.S.; Deng, L.; Wang, Y.; Dulubova, I.; Liu, X.; Rizo, J.; Südhof, T.C. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 2011, 144, 282–295.
- Deng, L.; Kaeser, P.S.; Xu, W.; Südhof, T.C. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 2011, 69, 317–331.
- Han, Y.; Kaeser, P.S.; Südhof, T.C.; Schneggenburger, R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 2011, 69, 304–316.
- Castillo, P.E.; Schoch, S.; Schmitz, F.; Südhof, T.C.; Malenka, R.C. RIM1α is required for presynaptic long-term potentiation. Nature 2002, 415, 327–330.
- Coppola, T.; Magnin-Lüthi, S.; Perret-Menoud, V.; Gattesco, S.; Schiavo, G.; Regazzi, R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J. Biol. Chem. 2001, 276, 32756–32762.
- Hibino, H.; Pironkova, R.; Onwumere, O.; Vologodskaia, M.; Hudspeth, A.J.; Lesage, F. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca2+ channels. Neuron 2002, 34, 411–423.
- Acuna, C.; Liu, X.; Gonzalez, A.; Südhof, T.C. RIM-BPs Mediate tight coupling of action potentials to Ca2+-triggered neurotransmitter release. Neuron 2015, 87, 1234–1247.
- Kiyonaka, S.; Wakamori, M.; Miki, T.; Uriu, Y.; Nonaka, M.; Bito, H.; Beedle, A.M.; Mori, E.; Hara, Y.; De Waard, M.; et al. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat. Neurosci 2007, 10, 691–701.
- Kiyonaka, S.; Nakajima, H.; Takada, Y.; Hida, Y.; Yoshioka, T.; Hagiwara, A.; Kitajima, I.; Mori, Y.; Ohtsuka, T. Physical and functional interaction of the active zone protein CAST/ERC2 and the β-subunit of the voltage-dependent Ca2+ channel. J. Biochem. 2012, 152, 149–159.
- Calloway, N.; Gouzer, G.; Xue, M.; Ryan, T.A. The active-zone protein Munc13 controls the use-dependence of presynaptic voltage-gated calcium channels. Elife 2015, 4, 1–15.
- Sudhof, T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci 2004, 27, 509–547.
- Cohen, M.W.; Jones, O.T.; Angelides, K.J. Distribution of Ca2+ channels on frog motor nerve terminals revealed by fluorescent omega-conotoxin. J. Neurosci. 1991, 11, 1032–1039.
- Westenbroek, R.E.; Hell, J.W.; Warner, C.; Dubel, S.J.; Snutch, T.P.; Catterall, W.A. Biochemical properties and subcellular distribution of an N-type calcium channel α1 subunit. Neuron 1992, 9, 1099–1115.
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418.
- Bennett, M.K.; Calakos, N.; Scheller, R.H. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 1992, 257, 255–259.
- Leveque, C.; el Far, O.U.S.S.A.M.A.; Martin-Moutot, N.; Sato, K.; Kato, R.; Takahashi, M.; Seagar, M.J. Purification of the N-type calcium channel associated with syntaxin and synaptotagmin. A complex implicated in synaptic vesicle exocytosis. J. Biol. Chem. 1994, 269, 6306–6312.
- Yoshida, A.; Oho, C.; Omori, A.; Kuwahara, R.; Ito, T.; Takahashi, M. HPC-1 is associated with synaptotagmin and ω-conotoxin receptor. J. Biol. Chem. 1992, 267, 24925–24928.
- Sheng, Z.H.; Rettig, J.; Cook, T.; Catterall, W.A. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature 1996, 379, 451–454.
- Sheng, Z.H.; Rettig, J.; Takahashi, M.; Catterall, W.A. Identification of a syntaxin-binding site on N-type calcium channels. Neuron 1994, 13, 1303–1313.
- Yokoyama, C.T.; Myers, S.J.; Fu, J.; Mockus, S.M.; Scheuer, T.; Catterall, W.A. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol. Cell Neurosci. 2005, 28, 1–17.
- Kim, D.K.; Catterall, W.A. Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 14782–14786.
- Rettig, J.; Sheng, Z.H.; Kim, D.K.; Hodson, C.D.; Snutch, T.P.; Catterall, W.A. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc. Natl. Acad. Sci. USA 1996, 93, 7363–7368.
- Bezprozvanny, I.; Scheller, R.H.; Tsien, R.W. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature 1995, 378, 623–626.
- Wiser, O.; Bennett, M.K.; Atlas, D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. Embo J. 1996, 15, 4100–4110.
- Zhong, H.; Yokoyama, C.T.; Scheuer, T.; Catterall, W.A. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat. Neurosci. 1999, 2, 939–941.
- Jarvis, S.E.; Zamponi, G.W. Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. J. Neurosci. 2001, 21, 2939–2948.
- Bezprozvanny, I.; Zhong, P.; Scheller, R.H.; Tsien, R.W. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc. Natl. Acad. Sci. USA 2000, 97, 13943–13948.
- Geppert, M.; Goda, Y.; Hammer, R.E.; Li, C.; Rosahl, T.W.; Stevens, C.F.; Südhof, T.C. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79, 717–727.
- Xu, J.; Mashimo, T.; Sudhof, T.C. Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 2007, 54, 567–581.
- Sheng, Z.H.; Yokoyama, C.T.; Catterall, W.A. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc. Natl. Acad. Sci. USA 1997, 94, 5405–5410.
- Wiser, O.; Tobi, D.; Trus, M.; Atlas, D. Synaptotagmin restores kinetic properties of a syntaxin-associated N-type voltage sensitive calcium channel. FEBS Lett. 1997, 404, 203–207.
- Yokoyama, C.T.; Sheng, Z.H.; Catterall, W.A. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J. Neurosci. 1997, 17, 6929–6938.
- DeMaria, C.D.; Soong, T.W.; Alseikhan, B.A.; Alvania, R.S.; Yue, D.T. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 2001, 411, 484–489.
- Lee, A.; Scheuer, T.; Catterall, W.A. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 2000, 20, 6830–6838.
- Lee, A.; Wong, S.T.; Gallagher, D.; Li, B.; Storm, D.R.; Scheuer, T.; Catterall, W.A. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 1999, 399, 155–159.
- Lee, A.; Zhou, H.; Scheuer, T.; Catterall, W.A. Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc. Natl. Acad. Sci. USA 2003, 100, 16059–16064.
- Few, A.P.; Lautermilch, N.J.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J. Neurosci. 2005, 25, 7071–7080.
- Lautermilch, N.J.; Few, A.P.; Scheuer, T.; Catterall, W.A. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J. Neurosci. 2005, 25, 7062–7070.
- Lee, A.; Westenbroek, R.E.; Haeseleer, F.; Palczewski, K.; Scheuer, T.; Catterall, W.A. Differential modulation of CaV2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 2002, 5, 210–217.
- Yan, J.; Leal, K.; Magupalli, V.G.; Nanou, E.; Martinez, G.Q.; Scheuer, T.; Catterall, W.A. Modulation of CaV2.1 channels by neuronal calcium sensor-1 induces short-term synaptic facilitation. Mol. Cell Neurosci. 2014, 63, 124–131.
- Liu, H.; De Waard, M.; Scott, V.E.; Gurnett, C.A.; Lennon, V.A.; Campbell, K.P. Identification of three subunits of the high affinity omega-conotoxin MVIIC-sensitive Ca2+ channel. J. Biol. Chem. 1996, 271, 13804–13810.
- Forsythe, I.D.; Tsujimoto, T.; Barnes-Davies, M.; Cuttle, M.F.; Takahashi, T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 1998, 20, 797–807.
- Xu, J.; Wu, L.G. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron 2005, 46, 633–645.
- Chaudhuri, D.; Alseikhan, B.A.; Chang, S.Y.; Soong, T.W.; Yue, D.T. Developmental activation of calmodulin-dependent facilitation of cerebellar P-type Ca2+ current. J. Neurosci. 2005, 25, 8282–8294.
- Mochida, S.; Few, A.P.; Scheuer, T.; Catterall, W.A. Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron 2008, 57, 210–216.
- Mochida, S.; Sheng, Z.H.; Baker, C.; Kobayashi, H.; Catterall, W.A. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron 1996, 17, 781–788.
- Rettig, J.; Heinemann, C.; Ashery, U.; Sheng, Z.H.; Yokoyama, C.T.; Catterall, W.A.; Neher, E. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J. Neurosci. 1997, 17, 6647–6656.
- Inchauspe, C.G.; Forsythe, I.D.; Uchitel, O.D. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J. Physiol. 2007, 584, 835–851.
- Iwasaki, S.; Momiyama, A.; Uchitel, O.D.; Takahashi, T. Developmental changes in calcium channel types mediating central synaptic transmission. J. Neurosci. 2000, 20, 59–65.
- Wu, L.G.; Borst, J.G. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron 1999, 23, 821–832.
- Wadel, K.; Neher, E.; Sakaba, T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron 2007, 53, 563–575.
- Wu, L.G.; Westenbroek, R.E.; Borst, J.G.G.; Catterall, W.A.; Sakmann, B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 1999, 19, 726–736.
- Keith, R.K.; Poage, R.E.; Yokoyama, C.T.; Catterall, W.A.; Meriney, S.D. Bidirectional modulation of transmitter release by calcium channel/syntaxin interactions in vivo. J. Neurosci. 2007, 27, 265–269.

