 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shogo Akahoshi | + 2203 word(s) | 2203 | 2020-12-22 07:54:58 | | | |
| 2 | Lily Guo | + 118 word(s) | 2321 | 2021-01-05 07:55:05 | | | | |
| 3 | Lily Guo | Meta information modification | 2321 | 2021-01-20 02:33:16 | | |
Video Upload Options
This entry focuses on steroid-induced adrenal insufficiency (SIAI) in children and discusses the latest findings by surveying recent studies. SIAI is a condition involving adrenocorticotropic hormone (ACTH) and cortisol suppression due to high doses or prolonged administration of glucocorticoids. While its chronic symptoms, such as fatigue and loss of appetite, are nonspecific, exposure to physical stressors, such as infection and surgery, increases the risk of adrenal crisis development accompanied by hypoglycemia, hypotension, or shock. The low-dose ACTH stimulation test is generally used for diagnosis, and the early morning serum cortisol level has also been shown to be useful in screening for the condition.
1. Introduction
1.1. Primary, Secondary, and Tertiary Adrenal Insufficiency
Adrenal insufficiency (AI) is defined as the inability of the adrenal cortex to produce sufficient amounts of glucocorticoid hormone. It can also be associated with mineralocorticoid deficiency, depending on the pathophysiology of the disease[1] Severe AI, or adrenal crisis, can be life-threatening because glucocorticoids and mineralocorticoids play a central role in maintaining energy, salt, and fluid homeostasis[2]. AI is usually classified into the primary and secondary types, which are caused by adrenal diseases and hypothalamic/pituitary diseases, respectively. AI associated with hypothalamic dysfunction is sometimes called tertiary AI. The main causes of AI are described below (see Table 1).
|
Primary Adrenal Insufficiency |
|
||
|
Autoimmune adrenalitis |
APS type 1, 2, 3 |
|
|
|
Infectious adrenalitis |
Tuberculosis, HIV-1, cryptococcosis, Treponema pallidum |
|
|
|
Bilateral adrenal hemorrhage |
Meningococcal sepsis |
|
|
|
Bilateral adrenal infiltration |
Primary adrenal lymphoma, hemochromatosis |
|
|
|
Drug-induced |
Ketoconazole, fluconazole, phenobarbital, phenytoin, rifampicin |
|
|
|
Genetic disorders |
CAH, adrenoleukodystrophy, adrenal hypoplasia congenita |
|
|
|
Secondary adrenal insufficiency |
|
||
|
Pituitary tumor |
Craniopharyngiomas, adenomas, cysts |
|
|
|
Pituitary injury |
Trauma, surgery, irradiation, pituitary apoplexy |
|
|
|
Pituitary infiltration |
Lymphocytic hypophysitis, tuberculosis, meningitis |
|
|
|
Genetic disorders |
Combined pituitary hormone deficiency, isolated ACTH deficiency |
|
|
|
Tertiary adrenal insufficiency |
|
||
|
Hypothalamic tumor |
Craniopharyngiomas, metastasis |
|
|
|
Hypothalamic injury |
Trauma, surgery, irradiation |
|
|
|
Hypothalamic infiltration |
Hemochromatosis, tuberculosis, meningitis |
|
|
|
Steroid-induced |
Systemic, inhalation, topical, intra-articular |
|
|
|
Other drug-induced |
Chlorpromazine, imipramine |
|
|
|
Abbreviations: ACTH, adrenocorticotropic hormone; APS, autoimmune polyendocrinopathy syndrome; CAH, congenital adrenal hyperplasia. |
|||
|
(Modified from [2] E. Charmandari, et al. Adrenal insufficiency. Lancet (London, England) 2014, 383, 2152–2167.) |
|
||
Primary AI can be further divided into the congenital type, represented by congenital adrenal hyperplasia (CAH), and the acquired type, represented by Addison’s disease[1]. One of the obvious differences in the clinical picture between primary, secondary, and tertiary AI is skin pigmentation, which is almost always present in primary AI (except in cases with a short duration) but is absent in secondary and tertiary AI [3]. Additionally, mineralocorticoid deficiency is generally associated with primary AI but not with secondary or tertiary AI because the renin–angiotensin–aldosterone system is not impaired in the latter[4]. In exceptional cases of CAH due to 11 beta-hydroxylase deficiency and 17 alpha-hydroxylase deficiency, mineralocorticoid excess occurs concurrently with glucocorticoid deficiency[5].
Secondary and tertiary AI can be divided into congenital and acquired types as well. Various genetic etiologies involving not only central nervous system malformations, such as holoprosencephaly, but also combined pituitary hormone deficiencies and isolated ACTH deficiency, have been identified[6]. Besides the drug-induced etiologies described below, acquired secondary AI can be caused by a tumor (e.g., craniopharyngioma), trauma, surgery, inflammation, or infarction involving the pituitary gland. It should be noted that secondary and tertiary AI may be latent or their onset may often be slow; congenital AI may become apparent around puberty (especially in septo-optic dysplasia[7] or hypopituitarism associated with perinatal problems[8]), while acquired AI can occur months to years after intracranial radiation[9] or traumatic brain injury[10][11]. Severe hypoglycemia in the neonatal period is rather exceptional and can be seen in congenital isolated ACTH deficiency caused by a TBX19 gene mutation[12].
1.2. Iatrogenic Adrenal Insufficiency
Iatrogenic AI refers to primary, secondary, or tertiary hypoadrenocorticism associated with drug administration, surgery, or irradiation. This review focuses on the role of steroids, the most frequent etiology of tertiary AI[2], which induce iatrogenic adrenal insufficiency (steroid-induced iatrogenic adrenal insufficiency, or SIAI) when the hypothalamic–pituitary–adrenal (HPA) axis is suppressed by high dosages or prolonged use of the drugs followed by abrupt discontinuation or rapid tapering. Patients with leukemia, asthma, collagen disease, or inflammatory bowel disease and those who have undergone transplant surgery[13] and have received long-term synthetic glucocorticoid treatment are especially at risk of SIAI development. There is currently insufficient evidence on the epidemiology, treatment strategy, and recovery process in SIAI; therefore, the optimal diagnostic criteria and management of the condition are still controversial. However, with the chief aim of preventing life-threatening adrenal crises[14], we discuss below the approaches available for managing patients treated with corticosteroids and summarize our tentative recommendations in Table 2. Other known causes of iatrogenic AI include drugs such as ketoconazole[15], mitotane [16] and etomidate[17], which inhibit the steroidogenesis pathway.
2. Clinical Manifestations of SIAI
2.1. Chronic Symptoms
Patients may present with chronic symptoms while recovering from HPA suppression. The common symptoms are weakness, fatigue, anorexia, and weight loss [18]. Frequent gastrointestinal complaints include nausea, vomiting, diarrhea, constipation, and abdominal pain, which are probably related to decreased bowel motility. These symptoms are more likely to occur with greater HPA suppression[1] and soon after stopping steroids [19]. In addition, adrenal insufficiency inevitably leads to a deficiency of dehydroepiandrosterone (DHEA), a substrate for peripheral sex hormone biosynthesis, which can lead to androgen deficiency in women. Its clinical manifestations include loss of axillary and pubic hair, dry skin, and decreased libido[20].
2.2. Symptoms as Side Effects of Glucocorticoids
Awareness of a variety of side effects of glucocorticoids is necessary when assessing SIAI. Particularly serious effects include immunosuppression, impaired growth, osteoporosis, and somewhat less frequently, but importantly, cataract formation and pancreatitis [21]. Medical Cushing’s syndrome, i.e., central obesity, muscle atrophy, and hypertension, can also occur in infants[22]. Nonspecific symptoms include mood disorders, abdominal symptoms, and dizziness[23], which can be difficult to differentiate from the symptoms of SIAI.
2.3. Acute Symptoms in the Presence of Stressors
It is even more important to note that physical stressors, such as severe infection or surgery, can trigger acute symptoms. Nonspecific symptoms, such as vomiting and diarrhea seen in Case 3, are common [18]. Laboratory findings are often normal [24]. Hyponatremia can develop as a result of increased vasopressin secretion and water retention[25]. The most severe and acute form is adrenal crisis, which consists of tachycardia, hypoglycemia, hypotension, dehydration, and acute abdominal pain [18][26]. Although the incidence of adrenal crisis is unknown, a questionnaire study of physicians across the UK revealed that adrenal crisis associated with inhaled corticosteroids (ICSs) occurred in 28 children, one of whom died from the condition. The mean patient age, duration of ICS treatment, and dosage of fluticasone, which was administered to 94% of the patients, was 6.4 years, 1.7 years, and 980 µg/day (range of 500–2000 µg/day), respectively[27]. At the time of the study, only 16% of all asthma patients were reportedly using fluticasone in the UK, indicating the gravity of the risk of adrenal crisis associated with fluticasone use[28].
3. Diagnostic Approaches to SIAI
3.1. Variation in Diagnostic Approaches
The symptoms of SIAI are nonspecific, and the diagnostic criteria are basically based on dynamic testing rather than the symptoms. The insulin tolerance test (ITT) has traditionally been the gold standard for HPA axis assessment in adults but this can be replaced by the safer, cheaper, and faster Synacthen test (SST)[29][30]. In pediatric medicine, the test most commonly used to diagnose secondary adrenal insufficiency is the low-dose ACTH stimulation test[31] because it is easy to administer, physiologically sound, safe, and reasonably sensitive. The glucagon [32] and CRH[14][33] stimulation tests (used in Cases 1–3 above) are also administered. Furthermore, early morning cortisol levels can also be used in screening[34][35] and assessment[36] of HPA function.
3.2. Low-Dose ACTH Stimulation Test
Low-dose ACTH stimulation testing is performed by administering 1 μg/1.73 m2 intravenous cosyntropin and assessing the subsequent increase in the serum cortisol level. A serum cortisol level of 16–20 μg/dL 30 min after cosyntropin administration is generally used as the threshold level although this may vary somewhat among assays[37][38][39][40]. Additional cortisol draws at 15 and 60 min may reduce the risk of false positive results[41]. We suggest performing this test at the initial evaluation 1 to 6 months after the end of the pharmacological dosing (depending on the duration of the steroid therapy), then every 3 to 6 months as needed in combination with early morning serum cortisol level testing, although the evidence and guideline recommendations supporting this approach are still inadequate. Once the values normalize, HDC maintenance therapy can be safely discontinued[19].
3.3. Early Morning Serum Cortisol Level
While random cortisol testing might be helpful in an emergency setting, as seen in Case 3, serum cortisol levels at 8 a.m. to 9 a.m. are often used when screening for SIAI. Previous studies have proposed an early morning cortisol level cutoff value of 8.5–10.3 µg/dL in adults[34][36]. A reference value of plasma cortisol, which is equivalent to that of serum cortisol[42][43][44], ≤3 μg/dL (83 nmol/L), may indicate SIAI, while a value >19 μg/dL (525 nmol/L) can be used to exclude SIAI[45]. A cohort study suggested a serum cortisol level of 5 µg/dL at 9 a.m. in children as a cutoff value for SIAI screening following steroid administration for Kawasaki disease[46].
4. Recovery Course in SIAI
4.1. Long-Term Administration
In an observational study of the natural history of SIAI in adults, both ACTH and cortisol secretion were initially suppressed. Then, ACTH secretion quickly increased before dropping to the normal range while cortisol secretion increased in response to the ACTH increase and normalized over several months[47]. Figure 1 shows a schema of ACTH and cortisol levels in SIAI following glucocorticoid therapy. SIAI due to steroid treatment for chronic diseases tends to show slower recovery. Studies reviewing adult cases of leukemia, hemangioma, and asthma reported that six to 12 months were required for recovery from SIAI[48], while a study of glomerular disease reported that 8.7 ± 4.6 months (mean ± SD)[35] were required. A pediatric study reported that recovery after treatment for acute lymphocytic leukemia took an average of 8.5 months (95% confidence interval: 6.3–10.7 months)[49], and another study reported that 11% of patients still had adrenal suppression 20 months after treatment for a rheumatic disease [50]. In line with the results of these studies, the patient in Case 2 did not show adequate cortisol secretion in the CRH stimulation test as late as eight months after the cessation of a six-week PSL treatment regimen.
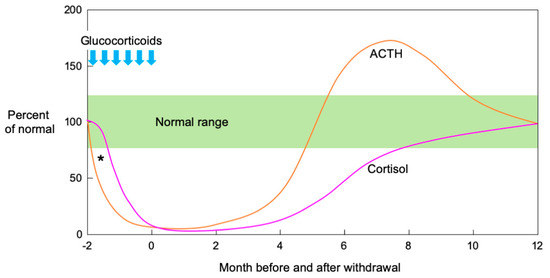
Figure 1. Schematic illustration of ACTH and cortisol levels after pharmacological glucocorticoid therapy. Both ACTH and cortisol secretion are initially suppressed when high-dose or long-term glucocorticoid administration is followed by abrupt cessation or rapid tapering. Theoretically, ACTH precedes cortisol in the suppression phase after pharmacological glucocorticoid therapy (*). In the recovery phase, ACTH secretion increases rapidly, then decreases to the normal range while cortisol secretion increases in response to increased ACTH and normalizes over several months. (Modified from[51] Kim E. Barrett, et al. The Adrenal Medulla & Adrenal Cortex. Ganong’s Review of Medical Physiology, 24th Ed. 2012, Chapter 20).
4.2. Short-Term Administration
If the dosages are small or the treatment short-term, recovery tends to be faster. The above literature review found that adrenal function recovered rapidly when steroids were administered for less than 14 days[48]. In an interventional study where a high-dose, short-term glucocorticoid (HDC 25 mg twice daily for five days) was administered to healthy adult subjects, the peak cortisol response to the ITT significantly decreased two days after PSL discontinuation but nearly returned to pretreatment levels five days after the intervention[52]. An observational study of childhood asthma found that adrenal function normalized in all 11 patients ten days after the completion of short-term PSL treatment[53], and another study of PSL treatment for Kawasaki disease in children reported recovery in more than half the patients within two months [46].
5. Practical Management of SIAI
5.1. Tapering from the Therapeutic to Physiological Replacement Dose
Weeks or months may be required to taper steroids until normal adrenal function is restored, and measures should be taken to prevent adrenal crisis during episodes of stress. In adults, the PSL dosage may be reduced from the pharmacological to the physiological level for several weeks. For example, depending on the patient’s condition, PSL may be reduced by 1 mg per day every two to four weeks[54]. Alternatively, HDC may be reduced weekly by 2.5 mg/day and maintained at 10 mg/day, which corresponds to endogenous cortisol synthesis as measured by isotope dilution[55].
In children, dose tapering to levels equivalent to the physiological dose (6–8 mg/m2/day of HDC[56][57][58]) is believed to be important to prevent symptoms of SIAI, although it has not been shown to restore adrenal cortical function[59]. Despite the lack of a consensus on how HDC should be prescribed, oral HDC administration once a day may be feasible and can improve compliance. Another option is to divide the total daily dose into four doses, two on waking, one at noon, and one in the evening, which may be acceptable to some patients[19].
5.2. Stress Doses
When the patient is under stress, the rescue method should be chosen according to the degree of the stress and the symptoms. Expert opinion [1][2] on steroid dosages for adults during stress was shown to be valid by a clinical study enrolling adults[60]. The stress dose for children is estimated by converting the adult dose into HDC per body surface area. For moderate physical stressors, such as infection associated with high fever (>38.5 °C), minor trauma, or dental treatment, three-fold the maintenance dosage or 50 mg/m2/day of HDC is administered orally in three to four divided doses [18][61]. If vomiting, lethargy, or other reasons make oral intake difficult, 50 mg/m2 HDC may first be injected intravenously. If an intravenous line cannot be placed, an intramuscular injection may be used[61]. In both adult and pediatric patients who are under severe stress, such as that caused by sepsis or major surgery, intravenous HDC 100 mg/m2/day administered continuously or every six hours in divided doses is recommended until recovery is achieved[18][26][61][62][63].
5.3. Patient Education
It is important that patients understand how to manage their condition, including knowing how much hydrocortisone to take or inject outside the hospital during episodes of stress. Patients are encouraged to wear a tag or a pin containing medical information, such as measures required in an emergency, hospital contact information, etc., which can enable others to provide assistance when needed [2][18][19].
References
- Oelkers, W. Adrenal insufficiency. N. Engl. J. Med. 1996, 335, 1206–1212, doi:10.1056/NEJM199610173351607.
- Charmandari, E.; Nicolaides, N.C.; Chrousos, G.P. Adrenal insufficiency. Lancet (Lond. Engl.) 2014, 383, 2152–2167, doi:10.1016/S0140-6736(13)61684-0.
- Crowley, R.K.; Argese, N.; Tomlinson, J.W.; Stewart, P.M. Central hypoadrenalism. J. Clin. Endocrinol. Metab. 2014, 99, 4027–4036, doi:10.1210/jc.2014-2476.
- Cooper, M.S.; Stewart, P.M. Corticosteroid insufficiency in acutely ill patients. N. Engl. J. Med. 2003, 348, 727–734, doi:10.1056/NEJMra020529.
- Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Yamakita, N. Endocrine causes of hypertension. Semin. Nephrol. 1995, 15, 106–115.
- Patti, G.; Guzzeti, C.; Di Iorgi, N.; Maria Allegri, A.E.; Napoli, F.; Loche, S.; Maghnie, M. Central adrenal insufficiency in children and adolescents. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 425–444, doi:10.1016/j.beem.2018.03.012.
- Cemeroglu, A.P.; Coulas, T.; Kleis, L. Spectrum of clinical presentations and endocrinological findings of patients with septo-optic dysplasia: A retrospective study. J. Pediatric Endocrinol. Metab. 2015, 28, 1057–1063, doi:10.1515/jpem-2015-0008.
- Miyamoto, J.; Hasegawa, Y.; Ohnami, N.; Onigata, K.; Kinoshita, E.; Nishi, Y.; Tachibana, K.; Hasegawa, T. Development of growth hormone and adrenocorticotropic hormone deficiencies in patients with prenatal or perinatal-onset hypothalamic hypopituitarism having invisible or thin pituitary stalk on magnetic resonance imaging. Endocr. J. 2001, 48, 355–362, doi:10.1507/endocrj.48.355.
- Oberfield, S.E.; Chin, D.; Uli, N.; David, R.; Sklar, C. Endocrine late effects of childhood cancers. J. Pediatrics 1997, 131, S37–S41, doi:10.1016/s0022-3476(97)70009-x.
- Benvenga, S.; Campenni, A.; Ruggeri, R.M.; Trimarchi, F. Clinical review 113: Hypopituitarism secondary to head trauma. J. Clin. Endocrinol. Metab. 2000, 85, 1353–1361, doi:10.1210/jcem.85.4.6506.
- Einaudi, S.; Bondone, C. The effects of head trauma on hypothalamic-pituitary function in children and adolescents. Curr. Opin. Pediatric 2007, 19, 465–470, doi:10.1097/MOP.0b013e3281ab6eeb.
- Peng, C.; Sun, G.; Tang, Z.; Hou, X. Congenital Isolated ACTH Deficiency Caused by TBX19 Gene Mutation: A Family Report. Front. Pediatric 2019, 7, 546, doi:10.3389/fped.2019.00546.
- Broersen, L.H.; Pereira, A.M.; Jorgensen, J.O.; Dekkers, O.M. Adrenal Insufficiency in Corticosteroids Use: Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2015, 100, 2171–2180, doi:10.1210/jc.2015-1218.
- Schlaghecke, R.; Kornely, E.; Santen, R.T.; Ridderskamp, P. The effect of long-term glucocorticoid therapy on pituitary-adrenal responses to exogenous corticotropin-releasing hormone. N. Engl. J. Med. 1992, 326, 226–230, doi:10.1056/NEJM199201233260403.
- Loose, D.S.; Kan, P.B.; Hirst, M.A.; Marcus, R.A.; Feldman, D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J. Clin. Investig. 1983, 71, 1495–1499, doi:10.1172/jci110903.
- Reimondo, G.; Puglisi, S.; Zaggia, B.; Basile, V.; Saba, L.; Perotti, P.; De Francia, S.; Volante, M.; Zatelli, M.C.; Cannavo, S., et al. Effects of mitotane on the hypothalamic-pituitary-adrenal axis in patients with adrenocortical carcinoma. Eur. J. Endocrinol. 2017, 177, 361–367, doi:10.1530/EJE-17-0452.
- Majesko, A.; Darby, J.M. Etomidate and adrenal insufficiency: The controversy continues. Crit. Care 2010, 14, 338, doi:10.1186/cc9338.
- Bornstein, S.R.; Allolio, B.; Arlt, W.; Barthel, A.; Don-Wauchope, A.; Hammer, G.D.; Husebye, E.S.; Merke, D.P.; Murad, M.H.; Stratakis, C.A., et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 364–389, doi:10.1210/jc.2015-1710.
- Barthel, A.; Willenberg, H.S.; Gruber, M.; Bornstein, S.R. Adrenal Insufficiency. Endocrinol. Adult Pediatric 2016, 102, 2704.
- Allolio, B.; Arlt, W. DHEA treatment: Myth or reality? Trends Endocrinol. Metab. 2002, 13, 288–294, doi:10.1016/s1043-2760(02)00617-3.
- Rimsza, M.E. Complications of corticosteroid therapy. Am. J. Dis. Child. 1978, 132, 806–810, doi:10.1001/archpedi.1978.02120330078018.
- Joshi, R.R.; Maresh, A. Iatrogenic Cushing's syndrome and adrenal insufficiency in infants on intranasal dexamethasone drops for nasal obstruction—Case series and literature review. Int. J. Pediatric Otorhinolaryngol. 2018, 105, 123–126, doi:10.1016/j.ijporl.2017.11.007.
- Klein-Gitelman, M.S.; Pachman, L.M. Intravenous corticosteroids: Adverse reactions are more variable than expected in children. J. Rheumatol. 1998, 25, 1995–2002.
- Aso, K.; Izawa, M.; Higuchi, A.; Kotoh, S.; Hasegawa, Y. Stress doses of glucocorticoids cannot prevent progression of all adrenal crises. Clin. Pediatric Endocrinol. 2009, 18, 23–27, doi:10.1297/cpe.18.23.
- Oelkers, W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N. Engl. J. Med. 1989, 321, 492–496, doi:10.1056/NEJM198908243210802.
- Kim, R.J.; Shah, R. Disorders of the Adrenal Gland. Netter Pediatrics 2011, 70, 864.
- Todd, G.R.; Acerini, C.L.; Ross-Russell, R.; Zahra, S.; Warner, J.T.; McCance, D. Survey of adrenal crisis associated with inhaled corticosteroids in the United Kingdom. Arch. Dis. Child. 2002, 87, 457–461, doi:10.1136/adc.87.6.457.
- Ahmet, A.; Kim, H.; Spier, S. Adrenal suppression: A practical guide to the screening and management of this under-recognized complication of inhaled corticosteroid therapy. Allergy Asthma Clin. Immunol. 2011, 7, 13, doi:10.1186/1710-1492-7-13.
- Lindholm, J.; Kehlet, H. Re-evaluation of the clinical value of the 30 min ACTH test in assessing the hypothalamic-pituitary-adrenocortical function. Clin. Endocrinol. (Oxf.) 1987, 26, 53–59, doi:10.1111/j.1365-2265.1987.tb03638.x.
- Stewart, P.M.; Corrie, J.; Seckl, J.R.; Edwards, C.R.; Padfield, P.L. A rational approach for assessing the hypothalamo-pituitary-adrenal axis. Lancet (Lond. Engl.) 1988, 1, 1208–1210, doi:10.1016/s0140-6736(88)92020-x.
- White, P.C. Adrenocortical Insufficiency. Nelson Textb. Pediatrics 2020, 593, 4264.
- Claahsen-van der Grinten, H.L.; Otten, B.J. Adrenal function: A gold standard test for adrenal insufficiency in children? Nat. Rev. Endocrinol. 2010, 6, 605–606, doi:10.1038/nrendo.2010.152.
- Kazlauskaite, R.; Maghnie, M. Pitfalls in the diagnosis of central adrenal insufficiency in children. Endocr. Dev. 2010, 17, 96–107, doi:10.1159/000262532.
- Montes-Villarreal, J.; Perez-Arredondo, L.A.; Rodriguez-Gutierrez, R.; Gonzalez-Colmenero, A.D.; Solis, R.C.; Gonzalez-Gonzalez, J.G.; Mancillas-Adame, L.G. Serum Morning Cortisol as a Screening Test for Adrenal Insufficiency. Endocr. Pract. 2020, 26, 30–35, doi:10.4158/EP-2019-0327.
- Karangizi, A.H.K.; Al-Shaghana, M.; Logan, S.; Criseno, S.; Webster, R.; Boelaert, K.; Hewins, P.; Harper, L. Glucocorticoid induced adrenal insufficiency is common in steroid treated glomerular diseases—proposed strategy for screening and management. BMC Nephrol. 2019, 20, 154, doi:10.1186/s12882-019-1354-6.
- Schmidt, I.L.; Lahner, H.; Mann, K.; Petersenn, S. Diagnosis of adrenal insufficiency: Evaluation of the corticotropin-releasing hormone test and Basal serum cortisol in comparison to the insulin tolerance test in patients with hypothalamic-pituitary-adrenal disease. J. Clin. Endocrinol. Metab. 2003, 88, 4193–4198, doi:10.1210/jc.2002-021897.
- Bowden, S.A.; Henry, R. Pediatric Adrenal Insufficiency: Diagnosis, Management, and New Therapies. Int. J. Pediatric 2018, 2018, 1739831, doi:10.1155/2018/1739831.
- Ospina, N.S.; Al Nofal, A.; Bancos, I.; Javed, A.; Benkhadra, K.; Kapoor, E.; Lteif, A.N.; Natt, N.; Murad, M.H. ACTH Stimulation Tests for the Diagnosis of Adrenal Insufficiency: Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2016, 101, 427–434, doi:10.1210/jc.2015-1700.
- Kazlauskaite, R.; Evans, A.T.; Villabona, C.V.; Abdu, T.A.; Ambrosi, B.; Atkinson, A.B.; Choi, C.H.; Clayton, R.N.; Courtney, C.H.; Gonc, E.N.; et al. Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: A metaanalysis. J. Clin. Endocrinol. Metab. 2008, 93, 4245–4253, doi:10.1210/jc.2008-0710.
- Dorin, R.I.; Qualls, C.R.; Crapo, L.M. Diagnosis of adrenal insufficiency. Ann. Intern. Med. 2003, 139, 194–204, doi:10.7326/0003-4819-139-3-200308050-00009.
- Gill, H.; Barrowman, N.; Webster, R.; Ahmet, A. Evaluating the Low-Dose ACTH Stimulation Test in Children: Ideal Times for Cortisol Measurement. J. Clin. Endocrinol. Metab. 2019, 104, 4587–4593, doi:10.1210/jc.2019-00295.
- SRL Inc. Serum Cortisol. Availabe online: http://test-guide.srl.info/hachioji/test/detail/004100902 (accessed on 25 November 2020).
- SRL Inc. Plasma Cortisol. Availabe online: http://test-guide.srl.info/hachioji/test/detail/004100906 (accessed on 25 November 2020).
- Siskos, A.P.; Jain, P.; Romisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665, doi:10.1021/acs.analchem.6b02930.
- Grinspoon, S.K.; Biller, B.M. Clinical review 62: Laboratory assessment of adrenal insufficiency. J. Clin. Endocrinol. Metab. 1994, 79, 923–931, doi:10.1210/jcem.79.4.7962298.
- Goto, M.; Miyagawa, N.; Kikunaga, K.; Miura, M.; Hasegawa, Y. Single serum cortisol values at 09:00 h can be indices of adrenocortical function in children with Kawasaki disease treated with intravenous immunoglobulin plus prednisolone. Clin. Pediatr Endocrinol. 2015, 24, 69–75, doi:10.1297/cpe.24.69.
- Graber, A.L.; Ney, R.L.; Nicholson, W.E.; Island, D.P.; Liddle, G.W. Natural History of Pituitary-Adrenal Recovery Following Long-Term Suppression with Corticosteroids. J. Clin. Endocrinol. Metab. 1965, 25, 11–16, doi:10.1210/jcem-25-1-11.
- Younes, A.K.; Younes, N.K. Recovery of steroid induced adrenal insufficiency. Transl. Pediatr 2017, 6, 269–273, doi:10.21037/tp.2017.10.01.
- Vestergaard, T.R.; Juul, A.; Lausten-Thomsen, U.; Lausen, B.; Hjalgrim, H.; Kvist, T.K.; Andersen, E.W.; Schmiegelow, K. Duration of adrenal insufficiency during treatment for childhood acute lymphoblastic leukemia. J. Pediatr Hematol. Oncol. 2011, 33, 442–449, doi:10.1097/MPH.0b013e3182260cbe.
- Huber, B.M.; Bolt, I.B.; Sauvain, M.J.; Fluck, C.E. Adrenal insufficiency after glucocorticoid withdrawal in children with rheumatic diseases. Acta Paediatr 2010, 99, 1889–1893, doi:10.1111/j.1651-2227.2010.01936.x.
- Kim, E. B.; Susan, M. B.; Scott, B.; Heddwen, L. B. The Adrenal Medulla & Adrenal Cortex. Ganong’s Review of Medical Physiology, 24th Ed. 2012, Chapter 20.
- Streck, W.F.; Lockwood, D.H. Pituitary adrenal recovery following short-term suppression with corticosteroids. Am. J. Med. 1979, 66, 910–914, doi:10.1016/0002-9343(79)90444-3.
- Zora, J.A.; Zimmerman, D.; Carey, T.L.; O'Connell, E.J.; Yunginger, J.W. Hypothalamic-pituitary-adrenal axis suppression after short-term, high-dose glucocorticoid therapy in children with asthma. J. Allergy Clin. Immunol. 1986, 77, 9–13, doi:10.1016/0091-6749(86)90315-5.
- Newell-Price, J.D.C.; Auchus, R.J. The Adrenal Cortex. Williams Textb. Endocrinol. 2019, 15, 1792.
- Esteban, N.V.; Loughlin, T.; Yergey, A.L.; Zawadzki, J.K.; Booth, J.D.; Winterer, J.C.; Loriaux, D.L. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J. Clin. Endocrinol. Metab. 1991, 72, 39–45, doi:10.1210/jcem-72-1-39.
- Linder, B.L.; Esteban, N.V.; Yergey, A.L.; Winterer, J.C.; Loriaux, D.L.; Cassorla, F. Cortisol production rate in childhood and adolescence. J. Pediatrics 1990, 117, 892–896, doi:10.1016/s0022-3476(05)80128-3.
- Esteban, N.V.; Yergey, A.L. Cortisol production rates measured by liquid chromatography/mass spectrometry. Steroids 1990, 55, 152–158, doi:10.1016/0039-128x(90)90103-i.
- Kerrigan, J.R.; Veldhuis, J.D.; Leyo, S.A.; Iranmanesh, A.; Rogol, A.D. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J. Clin. Endocrinol. Metab. 1993, 76, 1505–1510, doi:10.1210/jcem.76.6.8501158.
- Ahmet, A.; Brienza, V.; Tran, A.; Lemieux, J.; Aglipay, M.; Barrowman, N.; Duffy, C.; Roth, J.; Jurencak, R. Frequency and Duration of Adrenal Suppression Following Glucocorticoid Therapy in Children With Rheumatic Diseases. Arthritis Care Res. (Hoboken) 2017, 69, 1224–1230, doi:10.1002/acr.23123.
- Prete, A.; Taylor, A.E.; Bancos, I.; Smith, D.J.; Foster, M.A.; Kohler, S.; Fazal-Sanderson, V.; Komninos, J.; O'Neil, D.M.; Vassiliadi, D.A.; et al. Prevention of Adrenal Crisis: Cortisol Responses to Major Stress Compared to Stress Dose Hydrocortisone Delivery. J. Clin. Endocrinol. Metab. 2020, 105, doi:10.1210/clinem/dgaa133.
- Mass Screening, C.; Japanese Society for Pediatric, E.; Japanese Society for Mass, S.; Ishii, T.; Anzo, M.; Adachi, M.; Onigata, K.; Kusuda, S.; Nagasaki, K.; Harada, S.; et al. Guidelines for diagnosis and treatment of 21-hydroxylase deficiency (2014 revision). Clin. Pediatric Endocrinol. 2015, 24, 77–105, doi:10.1297/cpe.24.77.
- Shulman, D.I.; Palmert, M.R.; Kemp, S.F.; Lawson Wilkins, D.; Therapeutics, C. Adrenal insufficiency: Still a cause of morbidity and death in childhood. Pediatrics 2007, 119, e484–e494, doi:10.1542/peds.2006-1612.
- Woodcock, T.; Barker, P.; Daniel, S.; Fletcher, S.; Wass, J.A.H.; Tomlinson, J.W.; Misra, U.; Dattani, M.; Arlt, W.; Vercueil, A. Guidelines for the management of glucocorticoids during the peri-operative period for patients with adrenal insufficiency: Guidelines from the Association of Anaesthetists, the Royal College of Physicians and the Society for Endocrinology UK. Anaesthesia 2020, 75, 654–663, doi:10.1111/anae.14963.


