 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oxana Galzitskaya | + 3748 word(s) | 3748 | 2020-12-21 07:07:14 | | | |
| 2 | Stanislav Rimaso Kurpe | Meta information modification | 3748 | 2020-12-28 11:14:49 | | | | |
| 3 | Stanislav Rimaso Kurpe | Meta information modification | 3748 | 2020-12-28 11:55:19 | | | | |
| 4 | Nora Tang | Meta information modification | 3748 | 2020-12-29 04:18:20 | | | | |
| 5 | Oxana Galzitskaya | Meta information modification | 3748 | 2020-12-30 14:09:39 | | |
Video Upload Options
Theoretical methods for finding and predicting new antimicrobial peptides (AMPs), based on the use of specially designed programs for these purposes, are making an increasing contribution to the development of new AMPs. Machine learning methods are also used for the prediction. Artificial neural networks perform highly accurate predictions based on rules from databases of antimicrobial peptides. Recently published data on the development of new AMP drugs based on a combination of molecular design and genetic engineering approaches are presented. This review examines AMP development strategies from the perspective of the current high prevalence of antibiotic-resistant bacteria, and the potential prospects and challenges of using AMPs against infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). In turn, we proposed another strategy for the development of new AMPs based on predicting amyloidogenic regions in a protein molecule sequence.
1. Types of Programs for Prediction and Development of New AMPs
Over the past decades, great efforts have been made to discover AMPs in natural sources using wet biology techniques. Classical chromatographic methods for the detection of natural AMPs have played an important role in shaping the modern view of AMP in terms of biological source, amino acid sequence, three-dimensional structure, and antimicrobial activity. Since the 90s of the twentieth century, there are more and more possibilities for predicting and designing AMP with a specific function, pharmacological target and activity. The first report of such a discovery was associated with human cathelicidin, this peptide was detected using the method of the conservative sequence alignment of the pro-peptide domain [1]. In 2004, Yount and Yeaman discovered a motive for identifying new AMPs [2]. In addition, more efficient genomic and proteomic methods for the identification of peptides with antimicrobial function have been developed for peptides in recent years [3][4]. The application of prediction methods to genomes and proteomes, followed by experimental verification, accelerates the rate of discovery of peptides, which, in turn, allows expanding understanding of the functional nature of these compounds.
The similarity of peptides with and without antimicrobial activity is a bottleneck for bioinformatics methods. To solve this problem, programs for predicting antimicrobial activity based on various mathematic algorithms and information sources are used.
The programs of the first category use only data on the primary structure of mature AMPs. Based on these data, the G-X-C motif was discovered in peptides containing disulfide bonds (defensins) [5]. Using this motif, Yount and Yeaman discovered previously unknown peptides brazzein and charybdotoxin, which are active against bacteria and fungi Candida albicans [2]. A total of 28 new human defensins was identified. Programs in this category determine the length of the peptide, its charge, the content of hydrophobic residues in the molecule, and its amino acid composition. The predictions are made according to the correspondence of these peptide parameters to the range of parameters from the database.
The programs of the second category use highly conserved sequences of amino acid residues of pro-peptides. The amino acid sequences of mature peptides are more variable than the sequences of pro-peptides. This observation lies in the strategy of searching for such peptides [6]. Since the discovery of the first (antimicrobial) peptide, cathelicidin, in 1988, more than 100 such peptides have been identified. Analysis of processing signals allows predicting antimicrobial activity of other classes of AMPs. For example, amphibian antimicrobial peptide precursors also have a common and highly conserved pro-region, usually terminating in a typical Lys-Arg processing signal. This discovery was used as the basis for identifying many amphibian peptides [4][7].
A combination of approaches from previous types of programs is used in the third category. Fjell et al. created a program for prediction based on both pro-peptide sequences and mature peptides [8]. Peptides from Antimicrobial Sequences Database (AMSDb) are classified into several clusters, after which prediction parameters are calculated [9]. The authors identified 146 clusters for mature peptides and 40 clusters for pro-peptides. Using this program, it was possible to achieve 99% prediction accuracy. However, it is unclear how many more clusters can be found based on the most recent collection of naturally occurring AMPs [10], and whether it is possible to make the prediction even better.
The fourth type of programs is based on the search for homologous sequences of enzymes that carry out the transfer or modification of AMPs. LanM is a group of proteins that modify peptides from the lantibiotics family. The search for homologs of this group of proteins led to the discovery of new peptides in this family—haloduracin and lichenicidin [11]. Using this approach, 89 LanM homologues were identified; some of them were found in 61 strains that do not produce lantibiotics. The conserved LanT transporter was used to detect other lantibiotics. Based on the similarity with this protein, Singh and Sareen identified 54 bacterial strains containing LanT homologues [12]. Morton et al. used a protein cluster required for modification, transport, and resistance to bacteriocins for prediction [13]. Using this program, the authors predicted bacteriocin gene blocks for 2773 genomes.
The last type of program uses genetic information about expression, processing, and transport, and also performs comparisons with the already described AMPs. The processing of eukaryotic transcription data using data mining tools has allowed the identification of new AMPs. Lynn et al. identified 9 novel chicken AMPs by searching for homology tags of clustered expressed sequences using BLAST and a hidden Markov model [14]. Amaral et al. identified about a hundred AMPs based on physicochemical characteristics: peptide length, total charge surface and hydrophobic moment [15]. Proteolytic cleavage of proteins can release AMP. Based on this fact, Torrent et al. developed a theoretical method for identifying potentially active regions with antimicrobial activity [16]. Hellinger et al. achieved significant success by combining transcriptomic and proteomic data [17]. They were able to identify 164 AMPs from single plant: 108 based on transcriptomic data only and 127 based on mass spectrometry data. Perhaps one of the most effective strategies for searching of natural candidates for AMPs is based on in silico methods of analysis of RNAs synthesized after immunization of host cells with bacteria [18].
It is important to note that in order to predict AMPs that will act by the mechanism of coaggregation with the target protein (Figure 1), it is first of all necessary to search for amyloidogenic regions in this protein [19][20]. Approaches that include algorithms to predict aggregation/amyloidogenic sites may be useful for this purpose [21][22][23][24]. In addition, information on specific folded/unfolded or rigidity/flexibility regions of the protein may be appropriate for the development of AMP based on the sequence of the target protein [25][26].
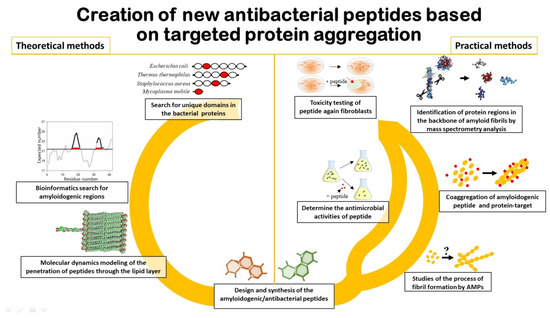
Figure 1. Schematic representation of the main aspects associated with the development of new antibacterial peptides.
It is important to take into account AMP development programs that are used to predict a possible immune response and allergic reactions to a specific peptide structure [27]. This validation is especially important for drug development based on AMPs, in particular, antiviral drugs, which will be discussed in more detail in the final section of this review.
2. SARS-CoV-2 Like an Object for Prediction and Development of New AMPs
Coronaviruses (CoVs) are a diverse group of viruses that infect a variety of animals, including live animals, including livestock, poultry, and can cause mild to severe respiratory infections in humans. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is a novel highly transmissible, pathogenic coronavirus that emerged at the end of 2019 and triggered a pandemic of new acute respiratory disease, also known as coronavirus disease 2019 (COVID-19). Previously, Severe Acute Respiratory Syndrome Coronavirus 1 (SARS-CoV-1) and Middle East Respiratory Syndrome Coronavirus (MERS-CoV) caused an outbreak of unusual viral pneumonia in 2002 and 2012, respectively [28]. 216 countries and regions from all six continents have reported more than 65 million COVID-19 cases, and more than 1.5 million patients have died, according to data from www.worldometer.info on December 4, 2020. The spread of SARS-CoV-2 is currently a serious threat to the health and life of people around the world. The efforts of scientists and specialists are aimed at developing effective methods of treating coronavirus infection. Clinical trials of various treatments for SARS-CoV-2 are currently underway, but none have been approved yet. Thus, SARS-CoV-2 is a relevant object for demonstrating the features of a strategy for the development of AMPs against a specific virus. It is well known that some peptides have antimicrobial activity against viruses. For example, melittin has significant antiviral activity and reduces the infectivity of enterovirus, human immunodeficiency virus (HIV), influenza A viruses, vesicular stomatitis virus (VSV), and some other viruses [29]. Thus, AMPs, derived from cathelicidin, effectively suppress Ebola virus the infection [30].
The genome of SARS-CoV-2 is around 30 kb, and is a positive-sense single-stranded RNA (+ssRNA) that contains six functional open reading frames (ORFs): spike (S), nucleocapsid (N), membrane (M), envelope (E), and replicase (ORF1a/ORF1b) [31]. The SARS-CoV-2 S protein consists of two subunits. The S1 subunit is important for binding to the host cell angiotensin-converting enzyme 2 (ACE2) receptor and the S2 subunit is responsible for the fusion of the virus and the cell membrane [32]. The S1 region is further divided into two functional domains, N-terminal domain (NTD) and three C-terminal domains (CTDs) [33]. CTDs contain a region (amino acids 319–529) of the receptor-binding domain (RBD), which plays a key role in contact with the hACE2 receptor [34]. Fourteen RBD residues bind to the hACE2 receptor. These are Tyr449, Tyr453, Asn487, Tyr489, Gly496, Thr500, Gly502, and Tyr505 are conserved in SARS-CoV-2 RBD, while Leu455, Phe456, Phe486, Gln493, Gln498 and Asn501 are substituted in different forms RBDs of SARS-CoV-1 and SARS-CoV-2 [35]. Biochemical analysis and pseudovirus penetration assay confirmed that the structural features of SARS-CoV-2 RBD have a higher hACE2 binding affinity than SARS-CoV RBDs [36]. The S2 subunit contains regions required for membrane fusion, including two heptad repeats (HR), a transmembrane domain (TM), an internal membrane fusion peptide (FP), and a membrane-proximal external region (MPER) [37].
Infection mechanisms have been investigated and it has been highlighted that the SARS-CoV-2 viral cell-surface spike protein targets hACE2 receptors. Cannalire et al. [38] discussed that the SBP1 peptide has been identified to inhibit the S/ACE2 interaction by targeting the RBD region of the S protein, but the RBD region is more prone to mutation, making it difficult to develop broad-spectrum inhibitors. In this case, the preferred approach is fusion inhibitors targeting the more stable heptad repeats (HRs) involved in the membrane fusion process. Among the peptides described, the EK1C4 lipopeptide exhibits strong S-mediated inhibition of fusion combined with selective broad antiviral activity in cellular assays against SARS-CoV-2 and other relevant CoVs such as MERS-CoV [38]. Another study used a programmatic approach to search and identification based on the hACE2 sequence of ten peptides that have a high potential for interaction with CoV-RBD [39]. Drugs targeting this mechanism are not yet available, but the recent work has demonstrated that peptide hACE2 mimics, creating from the H1 helix and consisting only of natural amino acids, block infection of human lung cells with IC50 in the nM range [40]. It is about eliminating the effect on the renin–angiotensin system and the kinin–kallikrein system in the light of the pathophysiological mechanisms associated with SARS-CoV-2 [41][42].
Similarly, to fusion proteins of other coronaviruses, SARS-CoV-2 S is activated by cellular proteases. It is assumed that the S1 and S2 subunits remain non-covalently linked after cleavage [43]. It is known that furin protease of the host cell cleaves the SARS-CoV-2 at the S1/S2 site. Cleavage at the S1/S2 site (residues 669−688) and subsequent cleavage at the S2’ site (residues 808−820) by the TMPRSS2 protease is important for virus penetration into lung cells [44][45]. Tavassoly et al. presented an interesting view that a peptide that is cut between two sites (S1/S2 and S2′) is detached from the virus and enters the intra- or extracellular environment. Subsequently, this peptide can induce some immunological reactions and act as a functional amyloid. The latter hypothesis was supported by the data of the AGGRESCAN program, which identified sites in the S-CoV peptide responsible for the self-aggregation properties [46]. Because the SARS-CoV-2 spike protein is critical for penetration into the host cell, it could be an interesting target protein option for the development of antiviral peptides. The identification of regions of a protein molecule prone to aggregation/formation of amyloid fibrils is an important step for the development of AMPs acting on the basis of a coaggregation mechanism. For this reason, we used several bioinformatics tools (FoldAmyloid, PASTA 2.0, Waltz and AGGRESCAN), which were previously used [19][47] to predict amyloidogenic sequences in proteins. The results of predicting amyloidogenic regions for the SARS-CoV-2 S protein by various methods are shown in Table 1.
Table 1. Amyloidogenic regions predicted by different programs for SARS-CoV-2 spike protein.
| Software | Amyloidogenic Regions for SARS-CoV-2 Spike Protein (https://www.uniprot.org/uniprot/P0DTC2) |
|---|---|
| AGGRESCAN (UAB, Barcelona, Spain http://bioinf.uab.es/aggrescan/) |
1–8, 10–16, 31–38, 40–47, 50–56, 58–70, 86–91, 100–109, 114–133, 140–145, 162–167, 189–208, 231–247, 260–271, 303–311, 331–336, 338–354, 362–383, 390–400, 428–437, 448–457, 484–490, 508–520, 584–588, 590–599, 610–617, 666–671, 688–698, 716–727, 729–745, 751–759, 761–768, 783–787, 797–805, 817–833, 853–861, 864–870, 872–914, 961–982, 1000–1013, 1044–1052, 1058–1068, 1097–1101, 1124–1137, 1171–1179, 1207–1249. |
| PASTA 2.0 (University of Padova, Padova, Italy http://protein.bio.unipd.it/pasta2/) |
1214–1244. |
| Waltz (Switch Laboratory, Leuven, Belgium http://waltz.switchlab.org/index.cgi) |
1–9, 88–95, 115–137, 141–146, 199–204, 261–271, 275–280, 365–378, 447–455, 485–496, 537–544, 608–618, 689–697, 703–727, 798–806, 913–940, 969–974, 1003–1015, 1063–1068, 1117–1122, 1175–1179, 1206–1233. |
| FoldAmyloid (Institute of Protein Research of the Russian Academy of Sciences, Pushchino, Russia http://bioinfo.protres.ru/fold-amyloid/) |
1–10, 34–38, 53–69,100–108,116–120, 126–135, 141–146, 174–178, 190–195, 235–244, 264–269, 274–278, 327–331, 348–353, 366–370, 389–395, 431– 436, 450–457, 487–492, 507–519, 539–543, 560–564, 582–586, 609–613, 691–697, 718–722, 738–743, 751– 755, 780–784, 799–804, 819–825, 875–880, 894–907, 993–999, 1005–1014, 1047–1051, 1060–1067, 1101–1105, 1127–1132, 1210–1239. |
As can be seen from Table 1, amyloidogenic regions identified by any software as amyloidogenic ones are predicted within one amino acid residue, but there are no completely similar predictions for FoldAmyloid, PASTA 2.0, Waltz and AGGRESCAN. However, the prediction of the amyloidogenic region of about 30 amino acid residues at the C-terminus of the SARS-CoV-2 spike protein, common for all four programs, is noteworthy. However, this region corresponds to the transmembrane domain. Interestingly, for the envelope (E) protein in SARS-CoV-1, amyloidogenic sequences at the C-terminus were also previously noted [48]. The same work discusses the possible role of the amyloidogenic sequence in the performance of protein E of its functions of binding to the host cell membranes and formation of ion channels. In our opinion, the amyloidogenic region in the middle of SARS-CoV-2 protein S can be the basis for the development of AMPs acting by the mechanism of directed coaggregation [49]. We hypothesize that anti-CoV peptides can bind to RBD of spike S1 protein (Figure 2). This interaction should reduce the binding affinity of the S1 to the hACE2 receptor of the target cell. A feature of this mechanism is the possibility of viral particles “sticking together”, which can prevent the spread of the virus and reduce the viral load.
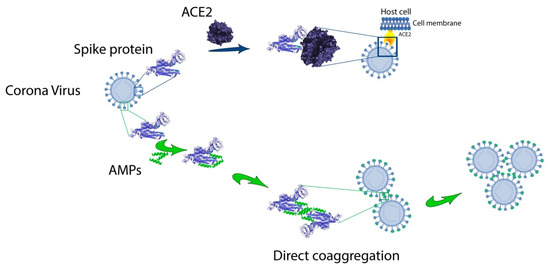
Figure 2. Hypothetical mechanism of direct coaggregation against CoVs.
Based on a similar logic, a number of authors tried to synthesize peptide inhibitors of the Spike–hACE2 interaction against SARS-CoV-1 and SARS-CoV-2 viruses [50][51][52][53][54]. As mentioned above, therapeutic peptides have several disadvantages: low bioavailability, short half-life and toxicity. Therefore, lipidation, PEGylation, glycosylation will be useful for improving the pharmacological properties of peptides. In addition, glycosylation can facilitate the specific recognition of viral particle binding sites. For example, how it happens, C-type lectins recognize pathogen patterns [55][56][57].
Recently, promising technologies for creating a vaccine against COVID-19 have been described. In these works, it is proposed to use antigenic peptides developed on the basis of peptide epitopes of viral proteins to inhibit SARS-CoV-2 infection and based on peptide epitopes of T and B lymphocytes to stimulate the human immune response [58]. High-performance in silico technologies currently allows the development of new approaches to the creation of peptide vaccines. Antigenic epitopes found in viral proteins can simultaneously meet several criteria, including immunodominant regions, non-allergenicity, population coverage, and lack of variability (conservatism) for efficient binding and molecular interaction with HLA (human leukocyte antigen) and TLR (toll-like receptor) alleles [27]. To evaluate the effectiveness of peptide vaccines against SARS-CoV-2, the frequencies of the HLA haplotypes can be used to predict the coverage of the developed vaccine. At the same time, peptides are evaluated based on the frequency of HLA haplotypes or HLA alleles in the target population, peptides with undesirable properties are filtered out, which are expected to be glycosylated, identical to peptides in the human proteome, or rapidly degraded [59]. Using in silico methods, epitopes of B cells, T cells, and IFN-gamma present on four structural proteins of the virus were mapped, and multi-epitope peptide-based vaccine was developed [60]. It should be taken into account when developing a peptide vaccine that some peptides can provoke an increased production of interleukin 6. In general, the increased mortality of patients with COVID-19 is associated with the induction of the cytokine storm. The work predicted 222 peptide sequences based on the viral spike protein, which can cause increased production of interleukin 6 (IL-6) [61]. The peptide protein kinase inhibitor CK2, previously proven in cancer, can also be used in the treatment of Covid-19. In a small (20 patient) randomized controlled clinical trial, CIGB-325 peptide reduced the mean number of lung lesions. However, in some patients, CIGB-325 may cause itching, redness, and rashes. In the future, it is planned to test the CIGB-325 peptide on more subjects in various combinations with antiviral drugs [62]. Preliminary encouraging results were obtained with the intravenous administration of the CIGB-258 immunomodulatory peptide to critically or severely ill COVID-19 patients. CIGB-258 significantly reduced the levels of biomarkers associated with hyperinflammation, IL-6 and tumor necrosis factor (TNFα) during treatment [63]. Another work suggests the use of annexin A1 Ac2-26 mimetic peptide, which reduces IL-6 production, pain and exudate, which could be a promising treatment in the fight against COVID-19 [64]. In addition, it is assumed that some short peptides, which that have already been developed and tested in the treatment of other diseases, can be used as inhibitors of the SARS-CoV-2 virus, as well as immunomodulators and bronchoprotectors of the pathological process with COVID-19 [65].
Drug redirection methods for the treatment of SARS-Cov-2 infection are currently being investigated. 300 peptide-like structures from various databases were selected. Using molecular dynamics modeling and docking analysis, the four peptide-like structures demonstrated strong binding affinity for amino acid residues within the SARS-CoV-2 site, the major protease of Mpro, also called 3CLpro [66][67]. The evolutionary aspect of SARS-CoV-2 should be considered in order to develop peptides that can further target the virus. Thus, in order to select four peptides with strong binding affinity for the main protease SARS-CoV-2, 2765 sequences containing a wide range of mutants from patients with COVID-19 were analyzed by molecular dynamics methods [68]. A computational approach was used to select 15 peptides that showed a higher affinity for RBD of SARS-CoV-2 S protein compared to the α-helix of the hACE2 receptor. It was noted that, in all the detected stable peptide-protein complexes, the Tyr489 and Tyr505 residues in RBD are involved in interaction, which suggests that they are critical for the binding of the discovered antiviral peptides to RBD [69]. After modeling based on the key interacting motifs of the spike protein, four new synthetic SARS-BLOCK™ peptides have been developed and characterized, which can serve as a combined therapeutic, immune and prophylactic agent against SARS-CoV-2. The technology may be interesting if it is possible to show the low cytotoxicity of the developed peptides [70].
Since the COVID-19 pandemic was announced, various options have been proposed to combat the infection. Recently, reviews have appeared that draw attention to the prospects for the development and use of antiviral peptides in the therapy of SARS-CoV-2, SARS-CoV-1, MERS-CoV and other respiratory viruses [71]. In particular, there was a point of view that synthetic or natural AMPs can be used to reduce the viral load on cells by blocking the contacts of the virus with receptors on the cell surface [72]. The use of protein/peptide inhalers, due to their prolonged action, higher efficacy, lower systemic availability and minimal toxicity, may be an effective approach for the treatment of SARS-CoV-2 [73]. Modification of existing natural AMPs is an attractive approach, since it can lead to the creation of new antiviral therapeutic agents [74]. The study shows the promise of using nisin, a dietary AMP produced by lactic acid bacteria, which can bind to hACE2, competitively inhibiting RBD SARS-CoV-2 [75]. Against COVID-19, it is proposed to use an AMP such as lactoferrin (LF), which is an iron-binding glycoprotein. However, it should be noted that the antiviral mechanisms of LF differ from virus to virus. Lactoferrin can enhance the host’s immunity against viral infection or bind directly to the viral particle or receptors and heparan sulfate proteoglycans on the cell-surface of the host cell [76][77]. Interestingly, a recent review suggests the use of peptide BPP-10c (Glu-Asn-Trp-Pro-His-Pro-Gln-Ile-Pro-Pro) derived from venom of Bothrops jararaca to counteract the effects of COVID-19 [78]. AMPs, which are found in the secretion of mesenchymal stem cells (MSCs), have antimicrobial properties, the ability to attenuate cytokine storms, and, therefore are an attractive approach to prevent the COVID-19 pandemic [79].
The influence of the coronavirus on the course of Alzheimer’s disease has been disclosed. It has been shown that Alzheimer’s disease can be triggered by various bacterial diseases, as well as certain viruses, albeit indirectly. The hypothesis is relevant due to the fact that many patients with COVID-19 have central nervous system disorders and cerebrovascular diseases. There is no data on the penetration of the virus into the human central nervous system, but this is important due to the presence of hACE2 expression in human nerve cells, and the virus can also enter the cerebrospinal fluid through the choroid plexus of the ventricles [80]. Interestingly, Aβ(1-42) peptide associated with Alzheimer’s disease may have antiviral and, in general, antimicrobial activity. AMPs have proven to be antiviral therapy that can be effective against infection and be used to reverse the effects of virus-induced inflammation. However, it is known that viral infections are often accompanied by bacterial infections. COVID-19 is no exception, and some patients die from bacterial co-infection. It can be assumed that the SARS-Cov-2 virus has a mechanism to suppress the production of the host’s own AMPs [81].
AMPs have immunomodulatory effects, with some activating and others suppressing inflammation, which should be considered when selecting candidates. To select specific immunogenic epitopes and accelerate vaccine development, the SARS-CoV-2 genes were analyzed for B and T cell epitope candidates. In addition, the authors propose to increase the low immunogenicity of a peptide vaccine by including an adjuvant in its composition and using an effective delivery system based on chitosan or a copolymer of lactic and glycolic acid (PLGA) [82]. It is suggested that peptides derived from the short-palate, lung and nasal epithelial clone-1 (SPLUNC1), alpha-1-antitrypsin (AAT), dornase alfa (DA) and neutralizing human S230 light chain antibodies can be used as anti-adhesion agents, preventing the SARS-CoV-2 virus from interacting with the human host cell. However, the use of the SPLUNC1 peptide may be associated with 33.3% of allergic reactions, based on in silico data [73]. It has been noted that clinical use of AMP may be hampered by general properties such as sensitivity to environmental conditions, large size, and poor distribution and excretion[83]. In the review, the authors discuss bioengineering strategies, chemical modifications, and combined approaches to finding solutions that improve the design, development of AMP, and peptide delivery technologies.
References
- B. Agerberth; H. Gunne; J. Odeberg; P. Kogner; H. G. Boman; G. H. Gudmundsson; FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis.. Proceedings of the National Academy of Sciences 1995, 92, 195-199, 10.1073/pnas.92.1.195.
- Nannette Y. Yount; Michael R. Yeaman; Multidimensional signatures in antimicrobial peptides. Proceedings of the National Academy of Sciences 2004, 101, 7363-7368, 10.1073/pnas.0401567101.
- Laurent Coquet; Jolanta Kolodziejek; Thierry Jouenne; Norbert Nowotny; Jay D. King; J. Michael Conlon; Peptidomic analysis of the extensive array of host-defense peptides in skin secretions of the dodecaploid frog Xenopus ruwenzoriensis (Pipidae). Comparative Biochemistry and Physiology Part D: Genomics and Proteomics 2016, 19, 18-24, 10.1016/j.cbd.2016.04.006.
- Xinwang Yang; Wen-Hui Lee; Yun Zhang; Extremely Abundant Antimicrobial Peptides Existed in the Skins of Nine Kinds of Chinese Odorous Frogs. Journal of Proteome Research 2011, 11, 306-319, 10.1021/pr200782u.
- Brian C. Schutte; Joseph P. Mitros; Jennifer A. Bartlett; Jesse D. Walters; Hong Peng Jia; Michael J. Welsh; Thomas L. Casavant; Paul B. McCray; Discovery of five conserved -defensin gene clusters using a computational search strategy. Proceedings of the National Academy of Sciences 2002, 99, 2129-2133, 10.1073/pnas.042692699.
- Margherita Zanetti; The role of cathelicidins in the innate host defenses of mammals.. Current Issues in Molecular Biology 2005, 7, 179-96.
- Yifeng Li; Xia Li; He Li; Oksana Lockridge; Guangshun Wang; A novel method for purifying recombinant human host defense cathelicidin LL-37 by utilizing its inherent property of aggregation. Protein Expression and Purification 2007, 54, 157-165, 10.1016/j.pep.2007.02.003.
- Christopher D. Fjell; Robert E.W. Hancock; Artem Cherkasov; AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148-1155, 10.1093/bioinformatics/btm068.
- Alessandro Tossi; Molecular Diversity in Gene-Encoded, Cationic Antimicrobial Polypeptides. Current Pharmaceutical Design 2002, 8, 743-761, 10.2174/1381612023395475.
- Guangshun Wang; Structural Analysis of Amphibian, Insect, and Plant Host Defense Peptides Inspires the Design of Novel Therapeutic Molecules. Host Defense Peptides and Their Potential as Therapeutic Agents 2016, 9, 229-252, 10.1007/978-3-319-32949-9_9.
- Maire Begley; Paul D. Cotter; Colin Hill; R. Ross; Identification of a Novel Two-Peptide Lantibiotic, Lichenicidin, following Rational Genome Mining for LanM Proteins. Applied and Environmental Microbiology 2009, 75, 5451-5460, 10.1128/aem.00730-09.
- Mangal Singh; Dipti Sareen; Novel LanT Associated Lantibiotic Clusters Identified by Genome Database Mining. PLOS ONE 2014, 9, e91352, 10.1371/journal.pone.0091352.
- James T. Morton; Stefan D. Freed; Shaun W. Lee; Iddo Friedberg; A large scale prediction of bacteriocin gene blocks suggests a wide functional spectrum for bacteriocins. BMC Bioinformatics 2015, 16, 1-9, 10.1186/s12859-015-0792-9.
- David J. Lynn; Rowan Higgs; Susan Gaines; Joanna Tierney; Tharappel James; Andrew T. Lloyd; Mario A. Fares; Grace Mulcahy; Cliona O’Farrelly; Bioinformatic discovery and initial characterisation of nine novel antimicrobial peptide genes in the chicken. Immunogenetics 2004, 56, 170-177, 10.1007/s00251-004-0675-0.
- André C. Amaral; Osmar N. Silva; Nathália C.C.R. Mundim; Maria J.A. De Carvalho; Ludovico Migliolo; Jose R.S.A. Leite; Maura V. Prates; Anamélia L. Bocca; Octávio L. Franco; Maria S.S. Felipe; et al. Predicting antimicrobial peptides from eukaryotic genomes: In silico strategies to develop antibiotics. Peptides 2012, 37, 301-308, 10.1016/j.peptides.2012.07.021.
- Marc Torrent; Victòria M. Nogués; Ester Boix; A theoretical approach to spot active regions in antimicrobial proteins. BMC Bioinformatics 2009, 10, 373-373, 10.1186/1471-2105-10-373.
- Roland Hellinger; Johannes Koehbach; Douglas E. Soltis; Eric J. Carpenter; Gane Ka-Shu Wong; Christian W Gruber; Peptidomics of Circular Cysteine-Rich Plant Peptides: Analysis of the Diversity of Cyclotides from Viola tricolor by Transcriptome and Proteome Mining. Journal of Proteome Research 2015, 14, 4851-4862, 10.1021/acs.jproteome.5b00681.
- Joon Ha Lee; Hoyong Chung; Yong Pyo Shin; In-Woo Kim; Sathishkumar Natarajan; Karpagam Veerappan; MinChul Seo; Junhyung Park; Jae Sam Hwang; Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction. Insects 2020, 11, 676, 10.3390/insects11100676.
- Sergei Yu. Grishin; Evgeniya I. Deryusheva; Andrey Machulin; Olga M. Selivanova; Anna V. Glyakina; Elena Yu. Gorbunova; Leila G. Mustaeva; Vyacheslav N. Azev; Valentina V. Rekstina; Tatyana S. Kalebina; et al.Alexey K. SurinOxana V. Galzitskaya Amyloidogenic Propensities of Ribosomal S1 Proteins: Bioinformatics Screening and Experimental Checking. International Journal of Molecular Sciences 2020, 21, 5199, 10.3390/ijms21155199.
- Oxana V. Galzitskaya; Sergiy O. Garbuzynskiy; Michail Yu. Lobanov; IS IT POSSIBLE TO PREDICT AMYLOIDOGENIC REGIONS FROM SEQUENCE ALONE?. Journal of Bioinformatics and Computational Biology 2006, 4, 373-388, 10.1142/s0219720006002004.
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S.; AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65, 10.1186/1471-2105-8-65.
- Sergiy O. Garbuzynskiy; Michail Yu. Lobanov; Oxana V. Galzitskaya; FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2009, 26, 326-332, 10.1093/bioinformatics/btp691.
- Ian Walsh; Flavio Seno; Silvio C E Tosatto; Antonio Trovato; PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Research 2014, 42, W301-W307, 10.1093/nar/gku399.
- Mikael Oliveberg; Waltz, an exciting new move in amyloid prediction. Nature Methods 2010, 7, 187-188, 10.1038/nmeth0310-187.
- O. V. Galzitskaya; S. O. Garbuzynskiy; M. Yu. Lobanov; Prediction of natively unfolded regions in protein chains. Molecular Biology 2006, 40, 298-304, 10.1134/s0026893306020166.
- Tatyana B. Mamonova; Anna V. Glyakina; Oxana V. Galzitskaya; Maria G. Kurnikova; Stability and rigidity/flexibility—Two sides of the same coin?. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013, 1834, 854-866, 10.1016/j.bbapap.2013.02.011.
- Niloofar Khairkhah; Mohammad Reza Aghasadeghi; Ali Namvar; Azam Bolhassani; Design of novel multiepitope constructs-based peptide vaccine against the structural S, N and M proteins of human COVID-19 using immunoinformatics analysis. PLoS ONE 2020, 15(10), e0240577, 10.1371/journal.pone.0240577.
- Peng Zhou; Xing-Lou Yang; Xian-Guang Wang; Ben Hu; Lei Zhang; Wei Zhang; Hao-Rui Si; Yan Zhu; Bei Li; Chao-Lin Huang; et al.Hui-Dong ChenJing ChenYun LuoHua GuoRen-Di JiangMei-Qin LiuYing ChenXu-Rui ShenXi WangXiao-Shuang ZhengKai ZhaoQuan-Jiao ChenFei DengLin-Lin LiuBing YanFa-Xian ZhanYan-Yi WangGeng-Fu XiaoZheng-Li Shi A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature Cell Biology 2020, 579, 270-273, 10.1038/s41586-020-2012-7.
- Hamed Memariani; Mojtaba Memariani; Hamideh Moravvej; Mohammad Shahidi-Dadras; Melittin: a venom-derived peptide with promising anti-viral properties. European Journal of Clinical Microbiology and Infections Diseases 2019, 39, 5-17, 10.1007/s10096-019-03674-0.
- Yangsheng Yu; Christopher L. Cooper; Guangshun Wang; M. Jane Morwitzer; Krishna Kota; Julie P. Tran; Steven B. Bradfute; Yan Liu; Jiayu Shao; Amanda K. Zhang; et al.Lindsey G. LuoSt. Patrick ReidSteven H. HinrichsKaihong Su Engineered Human Cathelicidin Antimicrobial Peptides Inhibit Ebola Virus Infection. iScience 2020, 23, 100999, 10.1016/j.isci.2020.100999.
- Yi Fan; Kai Zhao; Zhengli Shi; Peng Zhou; Bat Coronaviruses in China. Viruses 2019, 11, 210, 10.3390/v11030210.
- Yuan Huang; Chan Yang; Xin-Feng Xu; Wei Xu; Shuwen Liu; Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica 2020, 41, 1141-1149, 10.1038/s41401-020-0485-4.
- Jiahua He; Huanyu Tao; Yumeng Yan; Sheng-You Huang; Yi Xiao; Molecular Mechanism of Evolution and Human Infection with SARS-CoV-2. Viruses 2020, 12, 428, 10.3390/v12040428.
- Alexandra C. Walls; Young-Jun Park; M. Alejandra Tortorici; Abigail Wall; Andrew T. McGuire; David Veesler; Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281-292.e6, 10.1016/j.cell.2020.02.058.
- Amanat Ali; Ranjit Vijayan; Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Scientific Reports 2020, 10, 1-12, 10.1038/s41598-020-71188-3.
- Jian Shang; Yushun Wan; Chuming Luo; Gang Ye; Qibin Geng; Ashley Auerbach; Fang Li; Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences 2020, 117, 11727-11734, 10.1073/pnas.2003138117.
- Jinyong Zhang; Huazong Zeng; Jiang Gu; Haibo Li; Lixin Zheng; Quan M. Zou; Progress and Prospects on Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 153, 10.3390/vaccines8020153.
- Rolando Cannalire; Irina Stefanelli; Carmen Cerchia; Andrea Rosario Beccari; Sveva Pelliccia; Vincenzo Summa; SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. International Journal of Molecular Sciences 2020, 21, 5707, 10.3390/ijms21165707.
- Debmalya Barh; Sandeep Tiwari; Bruno Silva Andrade; Marta Giovanetti; Eduardo Almeida Costa; Ranjith Kumavath; Preetam Ghosh; Aristóteles Góes-Neto; Luiz Carlos Junior Alcantara; Vasco Azevedo; et al. Potential chimeric peptides to block the SARS-CoV-2 spike receptor-binding domain. F1000Research 2020, 9, 576, 10.12688/f1000research.24074.1.
- Philippe Karoyan; Vincent Vieillard; Estelle Odile; Alexis Denis; Luis Gómez-Morales; Pascal Grondin; Olivier Lequin; An hACE2 peptide mimic blocks SARS-CoV-2 Pulmonary Cell Infection. bioRxiv 2020, none, none, 10.1101/2020.08.24.264077.
- Roujian Lu; Xiang Zhao; Juan Li; Peihua Niu; Bo Yang; Honglong Wu; Wenling Wang; Hao Song; Baoying Huang; Na Zhu; et al.Yuhai BiXuejun MaFaxian ZhanLiang WangTao HuHong ZhouZhenhong HuWeimin ZhouLi ZhaoJing ChenYao MengJi WangYang LinJianying YuanZhihao XieJinmin MaWilliam J LiuDayan WangWenbo XuEdward C HolmesGeorge F GaoGuizhen WuWeijun ChenWeifeng ShiWenjie Tan Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020, 395, 565-574, 10.1016/s0140-6736(20)30251-8.
- Frank L. Van De Veerdonk; Mihai G Netea; Marcel Van Deuren; Jos Wm Van Der Meer; Quirijn De Mast; Roger J Brüggemann; Hans Van Der Hoeven; Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555, 10.7554/elife.57555.
- Sandrine Belouzard; Victor C. Chu; Gary R. Whittaker; Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proceedings of the National Academy of Sciences 2009, 106, 5871-5876, 10.1073/pnas.0809524106.
- Markus Hoffmann; Hannah Kleine-Weber; Stefan Pöhlmann; A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Molecular Cell 2020, 78, 779-784.e5, 10.1016/j.molcel.2020.04.022.
- Kristian G. Andersen; Andrew Rambaut; W. Ian Lipkin; Edward C Holmes; Robert F. Garry; The proximal origin of SARS-CoV-2. Nature Medicine 2020, 26, 450-452, 10.1038/s41591-020-0820-9.
- Omid Tavassoly; Farinaz Safavi; Iman Tavassoly; Seeding Brain Protein Aggregation by SARS-CoV-2 as a Possible Long-Term Complication of COVID-19 Infection. ACS Chemical Neuroscience 2020, 11, 3704-3706, 10.1021/acschemneuro.0c00676.
- Alexey K. Surin; Sergei Yu. Grishin; Oxana V. Galzitskaya; Identification of Amyloidogenic Regions in the Spine of Insulin Fibrils. Biochemistry (Moscow) 2019, 84, 47-55, 10.1134/s0006297919010061.
- Shruti Mukherjee; Dipita Bhattacharyya; Anirban Bhunia; Host-membrane interacting interface of the SARS coronavirus envelope protein: Immense functional potential of C-terminal domain. Biophysical Chemistry 2020, 266, 106452-106452, 10.1016/j.bpc.2020.106452.
- Stanislav R. Kurpe; Sergei Yu. Grishin; Alexey K. Surin; Olga M. Selivanova; Roman S. Fadeev; Ylyana F. Dzhus; Elena Yu. Gorbunova; Leila G. Mustaeva; Vyacheslav N. Azev; Oxana V. Galzitskaya; et al. Antimicrobial and Amyloidogenic Activity of Peptides Synthesized on the Basis of the Ribosomal S1 Protein from Thermus Thermophilus. International Journal of Molecular Sciences 2020, 21, 6382, 10.3390/ijms21176382.
- Shuai Xia; Yun Zhu; Meiqin Liu; Qiaoshuai Lan; Wei Xu; Yanling Wu; TianLei Ying; Shuwen Liu; Zhengli Shi; Shibo Jiang; et al.Lu Lu Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cellular & Molecular Immunology 2020, 17, 765-767, 10.1038/s41423-020-0374-2.
- Shuai Xia; Meiqin Liu; Chao Wang; Wei Xu; Qiaoshuai Lan; Siliang Feng; Feifei Qi; Linlin Bao; Lanying Du; Shuwen Liu; et al.Chuan QinFei SunZhengli ShiYun ZhuShibo JiangLu Lu Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Research 2020, 30, 343-355, 10.1038/s41422-020-0305-x.
- Kehu Yuan; Ling Yi; Jian Chen; Xiuxia Qu; Tingting Qing; Xi Rao; Pengfei Jiang; Jianhe Hu; Zikai Xiong; Yuchun Nie; et al.Xuanling ShiWei WangChen LingXiaolei YinKeqiang FanLuhua LaiMingxiao DingHongkui Deng Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochemical and Biophysical Research Communications 2004, 319, 746-752, 10.1016/j.bbrc.2004.05.046.
- Dong P. Han; Adam Penn-Nicholson; Michael W. Cho; Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15-25, 10.1016/j.virol.2006.01.029.
- Genwei Zhang; Sebastian Pomplun; Alexander Robert Loftis; Xuyu Tan; Andrei Loas; Bradley L. Pentelute; Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. bioRxiv 2020, none, none, 10.1101/2020.03.19.999318.
- Borong Lin; Xue Qing; Jinling Liao; Kan Zhuo; Role of Protein Glycosylation in Host-Pathogen Interaction. Cells 2020, 9, 1022, 10.3390/cells9041022.
- Anneke Engering; Teunis B. H. Geijtenbeek; Yvette Van Kooyk; Immune escape through C-type lectins on dendritic cells. Trends in Immunology 2002, 23, 480-485, 10.1016/s1471-4906(02)02296-2.
- Alessandra Cambi; Marjolein Koopman; Carl G. Figdor; How C-type lectins detect pathogens. Cellular Microbiology 2005, 7, 481-488, 10.1111/j.1462-5822.2005.00506.x.
- Murat Topuzoğullari; Tayfun Acar; Pelin Pelit Arayıcı; Burcu Uçar; Erennur Ugurel; Emrah Şefik Abamor; Tülin Arasoğlu; Dilek Turgut-Balik; Serap Derman; An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. TURKISH JOURNAL OF BIOLOGY 2020, 44, 215-227, 10.3906/biy-2006-1.
- Ge Liu; Brandon Carter; Trenton Bricken; Siddhartha Jain; Mathias Viard; Mary Carrington; David K. Gifford; Computationally Optimized SARS-CoV-2 MHC Class I and II Vaccine Formulations Predicted to Target Human Haplotype Distributions. Cell Systems 2020, 11, 131-144.e6, 10.1016/j.cels.2020.06.009.
- Abhishek Singh; Mukesh Thakur; Lalit Kumar Sharma; Kailash Chandra; Designing a multi-epitope peptide based vaccine against SARS-CoV-2. Scientific Reports 2020, 10, 1-12, 10.1038/s41598-020-73371-y.
- Anjali Dhall; Sumeet Patiyal; Neelam Sharma; Salman Sadullah Usmani; Gajendra P.S. Raghava; Computer-aided prediction and design of IL-6 inducing peptides: IL-6 plays a crucial role in COVID-19. Briefings in Bioinformatics 2020, 1, bbaa259, 10.1093/bib/bbaa259.
- Leticia R. Cruz; Idania Baladron; Aliusha Rittoles; Pablo A. Diaz; Carmen Valenzuela; Raul Santana; Maria M. Vazquez; Ariadna Garcia; Deyli Chacon; Delvin Thompson; et al. Treatment with an Anti-CK2 Synthetic Peptide Improves Clinical Response in Covid-19 Patients with Pneumonia. A Randomized and Controlled Clinical Trial. medRxiv 2020, none, none, 10.1101/2020.09.03.20187112.
- Maria Del Carmen Dominguez Horta Sr.; CIGB-258 immunomodulatory peptide: a novel promising treatment for critical and severe COVID-19 patients. medRxiv 2020, none, none, 10.1101/2020.05.27.20110601.
- Andre Gustavo Bonavita; Ac2-26 mimetic peptide of annexin A1 to treat severe COVID-19: A hypothesis. Medical Hypotheses 2020, 145, 110352-110352, 10.1016/j.mehy.2020.110352.
- Vladimir Khavinson; N. S. Linkova; Anastasiia Dyatlova; B. I. Kuznik; Roman Umnov; Peptides: Prospects for Use in the Treatment of COVID-19. Molecules 2020, 25, 4389, 10.3390/molecules25194389.
- Suyash Pant; Meenakshi Singh; V. Ravichandiran; U. S. N. Murty; Hemant Kumar Srivastava; Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics 2020, none, 1-10, 10.1080/07391102.2020.1757510.
- Linlin Zhang; Daizong Lin; Xinyuanyuan Sun; Ute Curth; Christian Drosten; Lucie Sauerhering; Stephan Becker; Katharina Rox; Rolf Hilgenfeld; Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409-412, 10.1126/science.abb3405.
- Ritika Kabra; Shailza Singh; Evolutionary Artificial Intelligence Based Peptide Discoveries for Effective Covid-19 Therapeutics. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020, 1867(1), 165978, 10.1016/j.bbadis.2020.165978.
- Surid Mohammad Chowdhury; Shafi Ahmad Talukder; Akib Mahmud Khan; Nadia Afrin; Ackas Ali; Rajib Islam; Rimon Parves; Abdulla Al Mamun; Abu Sufian; Nayeem Hossain; et al. Antiviral Peptides as Promising Therapeutics against SARS-CoV-2. The Journal of Physical Chemistry B 2020, 124, 9785-9792, 10.1021/acs.jpcb.0c05621.
- Andre Watson; Leonardo M R Ferreira; Peter Hwang; Jinbo Xu; Robert M. Stroud; Peptide Antidotes to SARS-CoV-2 (COVID-19). bioRxiv 2020, none, none, 10.1101/2020.08.06.238915.
- Arun Suria Karnan Mahendran; Yin Sze Lim; Chee-Mun Fang; Hwei-San Loh; Cheng Foh Le; The Potential of Antiviral Peptides as COVID-19 Therapeutics. Frontiers in Pharmacology 2020, 11, 575444, 10.3389/fphar.2020.575444.
- Biplab K. Maiti; Potential Role of Peptide-Based Antiviral Therapy Against SARS-CoV-2 Infection. ACS Pharmacology & Translational Science 2020, 3, 783-785, 10.1021/acsptsci.0c00081.
- Saad Salman; Fahad Hassan Shah; Maham Chaudhry; Muniba Tariq; Muhammad Yasir Akbar; Muhammad Adnan; In silico analysis of protein/peptide-based inhalers against SARS-CoV-2. Future Virology 2020, 15, 557-564, 10.2217/fvl-2020-0119.
- I-Ni Hsieh; Kevan L. Hartshorn; The Role of Antimicrobial Peptides in Influenza Virus Infection and Their Potential as Antiviral and Immunomodulatory Therapy. Pharmaceuticals 2016, 9, 53, 10.3390/ph9030053.
- Bhattacharya, R.; Gupta, A.M.; Mitra, S.; Mandal, S.; Biswas, S.R.; A natural food preservative peptide nisin can interact with the SARS-CoV-2 spike protein receptor human ACE2. Virology 2021, 552, 107–111, 10.1016/j.virol.2020.10.002.
- Sherif Elnagdy; Maha AlKhazindar; The Potential of Antimicrobial Peptides as an Antiviral Therapy against COVID-19. ACS Pharmacology & Translational Science 2020, 3, 780-782, 10.1021/acsptsci.0c00059.
- Jianshe Lang; Ning Yang; Jiejie Deng; Kangtai Liu; Peng Yang; Guigen Zhang; Chengyu Jiang; Inhibition of SARS Pseudovirus Cell Entry by Lactoferrin Binding to Heparan Sulfate Proteoglycans. PLOS ONE 2011, 6, e23710, 10.1371/journal.pone.0023710.
- Ahmed Salah Gouda; Bruno Mégarbane; Snake venom‐derived bradykinin‐potentiating peptides: A promising therapy for COVID ‐19?. Drug Development Research 2020, none, none, 10.1002/ddr.21732.
- Yogesh Kumar Verma; Ranjan Verma; Nishant Tyagi; AmanPreet Behl; Subodh Kumar; G. U. Gurudutta; COVID-19 and its Therapeutics: Special Emphasis on Mesenchymal Stem Cells Based Therapy. Stem Cell Reviews and Reports 2020, none, 1-19, 10.1007/s12015-020-10037-2.
- Giulia Abate; Maurizio Memo; Daniela Uberti; Impact of COVID-19 on Alzheimer’s Disease Risk: Viewpoint for Research Action. Healthcare 2020, 8, 286, 10.3390/healthcare8030286.
- Rasoul Mirzaei; Pedram Goodarzi; Muhammad Asadi; Ayda Soltani; Hussain Ali Abraham Aljanabi; Ali Salimi Jeda; Shirin Dashtbin; Saba Jalalifar; Rokhsareh Mohammadzadeh; Ali Teimoori; et al.Kamran TariMehdi SalariSima GhiasvandSima KazemiRasoul YousefimashoufHossein KeyvaniSajad Karampoor Bacterial co‐infections with SARS‐CoV ‐2. IUBMB Life 2020, 72, 2097-2111, 10.1002/iub.2356.
- Hui Xuan Lim; Jianhua Lim; Seyed Davoud Jazayeri; Sibrandes Poppema; Chit Laa Poh; Development of multi-epitope peptide-based vaccines against SARS-CoV-2. Biomedical Journal 2020, none, none, 10.1016/j.bj.2020.09.005.
- Huy Xuan Luong,Tung Truong Thanh, Tuan Hiep Tran; Antimicrobial peptides – Advances in development of therapeutic applications.. Life Sciences 2020, 260, 118407-118407, http://doi.org/10.1016/j.lfs.2020.118407.
- Huy Xuan Luong,Tung Truong Thanh, Tuan Hiep Tran; Antimicrobial peptides – Advances in development of therapeutic applications.. Life Sciences 2020, 260, 118407-118407, http://doi.org/10.1016/j.lfs.2020.118407.

