 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bani Medegan Fagla | -- | 5755 | 2024-03-28 20:50:40 | | | |
| 2 | Lindsay Dong | Meta information modification | 5755 | 2024-04-01 07:22:10 | | |
Video Upload Options
Protein misfolding disorders are a group of diseases characterized by supra-physiologic accumulation and aggregation of pathogenic proteoforms resulting from improper protein folding and/or insufficiency in clearance mechanisms. Although these processes have been historically linked to neurodegenerative disorders, such as Alzheimer’s disease, evidence linking protein misfolding to other pathologies continues to emerge. Indeed, the deposition of toxic protein aggregates in the form of oligomers or large amyloid fibrils has been linked to type 2 diabetes, various types of cancer, and, in more recent years, to preeclampsia, a life-threatening pregnancy-specific disorder. While extensive physiological mechanisms are in place to maintain proteostasis, processes, such as aging, genetic factors, or environmental stress in the form of hypoxia, nutrient deprivation or xenobiotic exposures can induce failure in these systems. As such, pregnancy, a natural physical state that already places the maternal body under significant physiological stress, creates an environment with a lower threshold for aberrant aggregation.
1. Introduction
2. The Central Hypothesis of PE Pathophysiology and Current Gaps
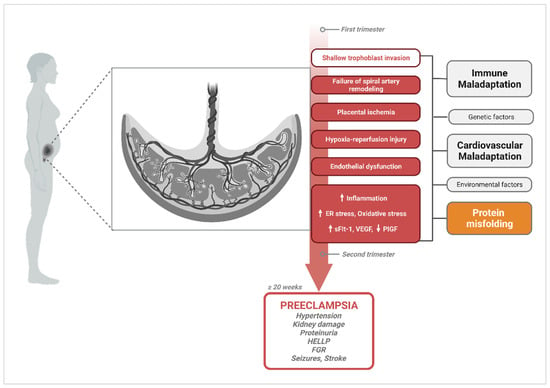
3. Protein Misfolding in PE
3.1. General Protein Misfolding and Aggregation Mechanisms
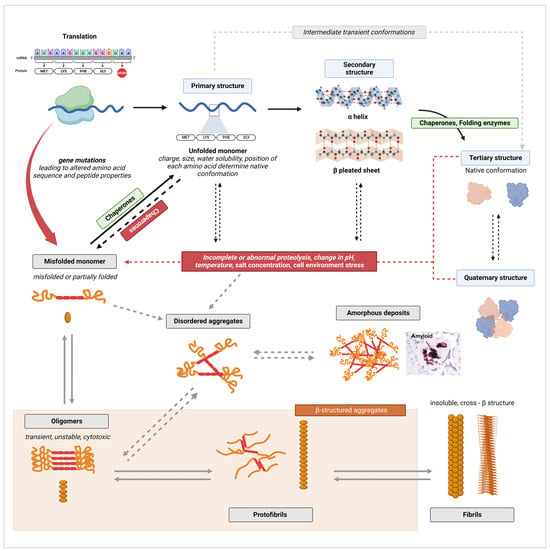
3.2. Excretion of Misfolded Proteins in the Urine
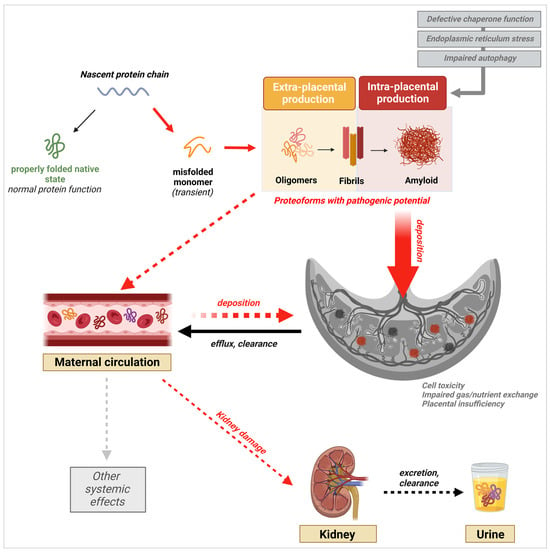
3.3. Presence of Misfolded Proteins in the Maternal Circulation
3.4. Placental Deposition of Extracellular Protein Aggregates
4. PE-Associated Misfoldome
4.1. SERPINA1/α-1-Antitrypsin (A1AT)
4.2. Aβ and Its Precursor APP
4.3. Tau
4.4. TTR
5. Driving Mechanisms of Protein Misfolding in PE
5.1. ER Stress and Hyperactivation of the Unfolded Protein Response (UPR)
5.2. Failure of the Protein Folding Machinery: The Role of Chaperones
5.3. Failure in Clearance Mechanisms: Defective Autophagolysosomal Processing
6. New Prediction, Diagnostic, and Treatment Approaches Based on PE-Associated Protein Misfolding
6.1. Early Prediction and Diagnosis
6.2. Treatment
7. Long-Term Implications of Pregnancy-Related Protein Misfolding Disease on Maternal and Fetal Health
References
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464.
- Bradbury, J. Chaperones: Keeping a Close Eye on Protein Folding. Lancet 2003, 361, 1194–1195.
- Chaudhuri, T.K.; Paul, S. Protein-Misfolding Diseases and Chaperone-Based Therapeutic Approaches. FEBS J. 2006, 273, 1331–1349.
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 2009, 78, 959–991.
- Williams, D. Pregnancy: A Stress Test for Life. Curr. Opin. Obstet. Gynecol. 2003, 15, 465.
- Williams, D.J. Physiological Changes of Normal Pregnancy. In Oxford Textbook of Medicine; Warrell, D.A., Cox, T.M., Firth, J.D., Eds.; Oxford University Press: Oxford, UK, 2010; ISBN 978-0-19-920485-4.
- Cunningham, F.G.; Leveno, K.J.; Bloom, S.L.; Dashe, J.S.; Hoffman, B.L.; Casey, B.M.; Spong, C.Y. Maternal Physiology. In Williams Obstetrics, 25e; McGraw-Hill Education: New York, NY, USA, 2018.
- Soma-Pillay, P.; Catherine, N.-P.; Tolppanen, H.; Mebazaa, A.; Tolppanen, H.; Mebazaa, A. Physiological Changes in Pregnancy. Cardiovasc. J. Afr. 2016, 27, 89–94.
- Kalhan, S.C. Protein Metabolism in Pregnancy. Am. J. Clin. Nutr. 2000, 71, 1249S–1255S.
- Duggleby, S.L.; Jackson, A.A. Protein, Amino Acid and Nitrogen Metabolism during Pregnancy: How Might the Mother Meet the Needs of Her Fetus? Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 503.
- Huppertz, B. Placental Origins of Preeclampsia. Hypertension 2008, 51, 970–975.
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-Eclampsia: Pathophysiology and Clinical Implications. BMJ 2019, 366, l2381.
- Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237.
- Copel, J.A.; Platt, L.D.; Hobbins, J.C.; Afshar, Y.; Grechukhina, O.; Mallampati, D.; Bromley, B.; Caughey, A.B.; Grobman, W.; Han, C.S.; et al. Gottesfeld-Hohler Memorial Foundation Risk Assessment for Early-Onset Preeclampsia in the United States: Think Tank Summary. Obstet. Gynecol. 2020, 135, 36.
- Reddy, M.; Fenn, S.; Rolnik, D.L.; Mol, B.W.; da Silva Costa, F.; Wallace, E.M.; Palmer, K.R. The Impact of the Definition of Preeclampsia on Disease Diagnosis and Outcomes: A Retrospective Cohort Study. Am. J. Obstet. Gynecol. 2021, 224, 217.e1–217.e11.
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-Eclampsia Part 1: Current Understanding of Its Pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466.
- Roberts, J.M.; Hubel, C.A. The Two Stage Model of Preeclampsia: Variations on the Theme. Placenta 2009, 30, S32–S37.
- Redman, C.W.G.; Sargent, I.L. REVIEW ARTICLE: Immunology of Pre-Eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543.
- Brosens, I.; Robertson, W.B.; Dixon, H.G. The Physiological Response of the Vessels of the Placental Bed to Normal Pregnancy. J. Pathol. Bacteriol. 1967, 93, 569–579.
- Pijnenborg, R.; Bland, J.M.; Robertson, W.B.; Brosens, I. Uteroplacental Arterial Changes Related to Interstitial Trophoblast Migration in Early Human Pregnancy. Placenta 1983, 4, 397–413.
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The Uterine Spiral Arteries in Human Pregnancy: Facts and Controversies. Placenta 2006, 27, 939–958.
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral Artery Remodeling and Trophoblast Invasion in Preeclampsia and Fetal Growth Restriction. Hypertension 2013, 62, 1046–1054.
- Hamilton, W.J.; Boyd, J.D. Trophoblast in Human Utero-Placental Arteries. Nature 1966, 212, 906–908.
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201.
- Burton, G.J.; Jauniaux, E. Placental Oxidative Stress: From Miscarriage to Preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352.
- Ball, E.; Bulmer, J.; Ayis, S.; Lyall, F.; Robson, S. Late sporadic miscarriage is associated with abnormalities in spiral artery transformation and trophoblast invasion. J. Pathol. 2006, 208, 535–542.
- Kim, Y.M.; Bujold, E.; Chaiworapongsa, T.; Gomez, R.; Yoon, B.H.; Thaler, H.T.; Rotmensch, S.; Romero, R. Failure of Physiologic Transformation of the Spiral Arteries in Patients with Preterm Labor and Intact Membranes. Am. J. Obstet. Gynecol. 2003, 189, 1063–1069.
- Khong, T.Y.; De Wolf, F.; Robertson, W.B.; Brosens, I. Inadequate Maternal Vascular Response to Placentation in Pregnancies Complicated by Pre-Eclampsia and by Small-for-Gestational Age Infants. Br. J. Obstet. Gynaecol. 1986, 93, 1049–1059.
- Veerbeek, J.H.W.; Nikkels, P.G.J.; Torrance, H.L.; Gravesteijn, J.; Post Uiterweer, E.D.; Derks, J.B.; Koenen, S.V.; Visser, G.H.A.; Van Rijn, B.B.; Franx, A. Placental Pathology in Early Intrauterine Growth Restriction Associated with Maternal Hypertension. Placenta 2014, 35, 696–701.
- Pathak, S.; Lees, C.C.; Hackett, G.; Jessop, F.; Sebire, N.J. Frequency and Clinical Significance of Placental Histological Lesions in an Unselected Population at or near Term. Virchows Arch. 2011, 459, 565–572.
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess Placental Soluble Fms-like Tyrosine Kinase 1 (sFlt1) May Contribute to Endothelial Dysfunction, Hypertension, and Proteinuria in Preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble Endoglin and Other Circulating Antiangiogenic Factors in Preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005.
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22.
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia—Pathophysiology and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702.
- Huppertz, B.; Sammar, M.; Chefetz, I.; Neumaier-Wagner, P.; Bartz, C.; Meiri, H. Longitudinal Determination of Serum Placental Protein 13 during Development of Preeclampsia. Fetal Diagn. Ther. 2008, 24, 230–236.
- Craven, C.M.; Morgan, T.; Ward, K. Decidual Spiral Artery Remodelling Begins before Cellular Interaction with Cytotrophoblasts. Placenta 1998, 19, 241–252.
- Vikse, B.E.; Irgens, L.M.; Leivestad, T.; Skjaerven, R.; Iversen, B.M. Preeclampsia and the Risk of End-Stage Renal Disease. N. Engl. J. Med. 2008, 359, 800–809.
- Kattah, A. Preeclampsia and Kidney Disease: Deciphering Cause and Effect. Curr. Hypertens. Rep. 2020, 22, 91.
- Wiles, K.; Chappell, L.C.; Lightstone, L.; Bramham, K. Updates in Diagnosis and Management of Preeclampsia in Women with CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 1371–1380.
- Hirose, N.; Ohkuchi, A.; Matsubara, S.; Suzuki, M. Risk of Preeclampsia in Women with CKD, Dialysis or Kidney Transplantation. Med. J. Obstet. Gynecol. 2014, 2, 1028.
- Kendrick, J.; Holmen, J.; You, Z.; Smits, G.; Chonchol, M. Association of Unilateral Renal Agenesis with Adverse Outcomes in Pregnancy: A Matched Cohort Study. Am. J. Kidney Dis. 2017, 70, 506–511.
- Dupont, V.; Berg, A.H.; Yamashita, M.; Huang, C.; Covarrubias, A.E.; Ali, S.; Stotland, A.; Van Eyk, J.E.; Jim, B.; Thadhani, R.; et al. Impaired Renal Reserve Contributes to Preeclampsia via the Kynurenine and Soluble Fms–like Tyrosine Kinase 1 Pathway. J. Clin. Investig. 2022, 132, e158346.
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68.
- Burton, G.J.; Yung, H.-W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta 2009, 30, 43–48.
- Saibil, H. Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642.
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 612757.
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2005, 1, 131–140.
- Anfinsen, C.B. Principles That Govern the Folding of Protein Chains. Science 1973, 181, 223–230.
- Dobson, C.M. The Structural Basis of Protein Folding and Its Links with Human Disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 133–145.
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890.
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396.
- Newberry, R.W.; Raines, R.T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14, 1677.
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155.
- Kang, T.S.; Kini, R.M. Structural Determinants of Protein Folding. Cell. Mol. Life Sci. 2009, 66, 2341–2361.
- Dobson, C.M. Principles of Protein Folding, Misfolding and Aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16.
- Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Protein Solubility and Protein Homeostasis: A Generic View of Protein Misfolding Disorders. Cold Spring Harb. Perspect. Biol. 2011, 3, a010454.
- Kelly, J.W. The Alternative Conformations of Amyloidogenic Proteins and Their Multi-Step Assembly Pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106.
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763.
- Knowles, T.P.J.; White, D.A.; Abate, A.R.; Agresti, J.J.; Cohen, S.I.A.; Sperling, R.A.; De Genst, E.J.; Dobson, C.M.; Weitz, D.A. Observation of Spatial Propagation of Amyloid Assembly from Single Nuclei. Proc. Natl. Acad. Sci. USA 2011, 108, 14746–14751.
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From Macroscopic Measurements to Microscopic Mechanisms of Protein Aggregation. J. Mol. Biol. 2012, 421, 160–171.
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537.
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610.
- Koike, H.; Iguchi, Y.; Sahashi, K.; Katsuno, M. Significance of Oligomeric and Fibrillar Species in Amyloidosis: Insights into Pathophysiology and Treatment. Molecules 2021, 26, 5091.
- Buhimschi, I.A.; Zhao, G.; Funai, E.F.; Harris, N.; Sasson, I.E.; Bernstein, I.M.; Saade, G.R.; Buhimschi, C.S. Proteomic profiling of urine identifies specific fragments of Serpina-1 and albumin as biomarkers of preeclampsia. Am. J. Obstet. Gynecol. 2008, 199, 551.e1–551.e16.
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein Misfolding, Congophilia, Oligomerization, and Defective Amyloid Processing in Preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92.
- Puchtler, H.; Sweat, F.; Levine, M. On the binding of congo red by amyloid. J. Histochem. Cytochem. 1962, 10, 355–364.
- Kalkunte, S.S.; Neubeck, S.; Norris, W.E.; Cheng, S.-B.; Kostadinov, S.; Vu Hoang, D.; Ahmed, A.; von Eggeling, F.; Shaikh, Z.; Padbury, J.; et al. Transthyretin Is Dysregulated in Preeclampsia, and Its Native Form Prevents the Onset of Disease in a Preclinical Mouse Model. Am. J. Pathol. 2013, 183, 1425–1436.
- Cheng, S.; Banerjee, S.; Daiello, L.A.; Nakashima, A.; Jash, S.; Huang, Z.; Drake, J.D.; Ernerudh, J.; Berg, G.; Padbury, J.; et al. Novel Blood Test for Early Biomarkers of Preeclampsia and Alzheimer’s Disease. Sci. Rep. 2021, 11, 15934.
- Carrell, R.W.; Mushunje, A.; Zhou, A. Serpins Show Structural Basis for Oligomer Toxicity and Amyloid Ubiquity. FEBS Lett. 2008, 582, 2537–2541.
- Lewandowski, C.T.; Weng, J.M.; LaDu, M.J. Alzheimer’s Disease Pathology in APOE Transgenic Mouse Models: The Who, What, When, Where, Why, and How. Neurobiol. Dis. 2020, 139, 104811.
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2010, 17, 514–524.
- Tong, M.; Cheng, S.; Chen, Q.; DeSousa, J.; Stone, P.R.; James, J.L.; Chamley, L.W.; Sharma, S. Aggregated Transthyretin Is Specifically Packaged into Placental Nano-Vesicles in Preeclampsia. Sci. Rep. 2017, 7, 6694.
- Roussel, B.D.; Irving, J.A.; Ekeowa, U.I.; Belorgey, D.; Haq, I.; Ordóñez, A.; Kruppa, A.J.; Duvoix, A.; Rashid, S.T.; Crowther, D.C.; et al. Unravelling the Twists and Turns of the Serpinopathies. FEBS J. 2011, 278, 3859–3867.
- Köhnlein, T.; Welte, T. Alpha-1 Antitrypsin Deficiency: Pathogenesis, Clinical Presentation, Diagnosis, and Treatment. Am. J. Med. 2008, 121, 3–9.
- Yuan, Y.; DiCiaccio, B.; Li, Y.; Elshikha, A.S.; Titov, D.; Brenner, B.; Seifer, L.; Pan, H.; Karic, N.; Akbar, M.A.; et al. Anti-inflammaging Effects of Human Alpha-1 Antitrypsin. Aging Cell 2018, 17, e12694.
- Starodubtseva, N.; Nizyaeva, N.; Baev, O.; Bugrova, A.; Gapaeva, M.; Muminova, K.; Kononikhin, A.; Frankevich, V.; Nikolaev, E.; Sukhikh, G. SERPINA1 Peptides in Urine as A Potential Marker of Preeclampsia Severity. Int. J. Mol. Sci. 2020, 21, 914.
- Cheng, S.-B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis Is a Critical Inflammatory Pathway in the Placenta from Early Onset Preeclampsia and in Human Trophoblasts Exposed to Hypoxia and Endoplasmic Reticulum Stressors. Cell Death Dis. 2019, 10, 927.
- Twina, G.; Sheiner, E.; Shahaf, G.; Yaniv Salem, S.; Madar, T.; Baron, J.; Wiznitzer, A.; Mazor, M.; Holcberg, G.; Lewis, E.C. Lower Circulation Levels and Activity of α-1 Antitrypsin in Pregnant Women with Severe Preeclampsia. J. Matern.-Fetal Neonatal Med. 2012, 25, 2667–2670.
- Feng, Y.; Xu, J.; Zhou, Q.; Wang, R.; Liu, N.; Wu, Y.; Yuan, H.; Che, H. Alpha-1 Antitrypsin Prevents the Development of Preeclampsia Through Suppression of Oxidative Stress. Front. Physiol. 2016, 7, 176.
- Feng, Y.; Wang, N.; Xu, J.; Zou, J.; Liang, X.; Liu, H.; Chen, Y. Alpha-1-Antitrypsin Functions as a Protective Factor in Preeclampsia through Activating Smad2 and Inhibitor of DNA Binding 4. Oncotarget 2017, 8, 113002–113012.
- Nagarajappa, C.; Rangappa, S.S.; Balakrishna, S. Misfolding Linked Mutations of SERPINA1 Gene Are Uncommon in Preeclampsia. Arch. Med. Health Sci. 2019, 7, 177.
- Shaik, N.A.; Saud Al-Saud, N.B.; Abdulhamid Aljuhani, T.; Jamil, K.; Alnuman, H.; Aljeaid, D.; Sultana, N.; El-Harouni, A.A.; Awan, Z.A.; Elango, R.; et al. Structural Characterization and Conformational Dynamics of Alpha-1 Antitrypsin Pathogenic Variants Causing Alpha-1-Antitrypsin Deficiency. Front. Mol. Biosci. 2022, 9, 1051511.
- Sergeeva, V.A.; Zakharova, N.V.; Bugrova, A.E.; Starodubtseva, N.L.; Indeykina, M.I.; Kononikhin, A.S.; Frankevich, V.E.; Nikolaev, E.N. The High-Resolution Mass Spectrometry Study of the Protein Composition of Amyloid-like Urine Aggregates Associated with Preeclampsia. Eur. J. Mass. Spectrom. 2020, 26, 158–161.
- Valtanen, R.S.; Buhimschi, C.S.; Bahtiyar, M.O.; Zhao, G.; Jing, H.; Ackerman, W.E.; Glabe, C.G.; Buhimschi, I.A. Conformation-Dependent Anti-Aβ Monoclonal Antibody Signatures of Disease Status and Severity in Urine of Women with Preeclampsia. Pregnancy Hypertens. 2022, 28, 51–59.
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The Key Peptide in the Pathogenesis of Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 221.
- Müller, U.C.; Deller, T.; Korte, M. Not Just Amyloid: Physiological Functions of the Amyloid Precursor Protein Family. Nat. Rev. Neurosci. 2017, 18, 281–298.
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235.
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent Developments in Clinical Biomarkers and Therapies Targeting Tau Phosphorylation in Alzheimer’s Disease and Other Tauopathies. Mol. Neurodegener. 2021, 16, 37.
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau Protein in Health and Disease. Acta Neuropathol. 2017, 133, 665–704.
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402.
- Patel, J.; Landers, K.A.; Li, H.; Mortimer, R.H.; Richard, K. Ontogenic Changes in Placental Transthyretin. Placenta 2011, 32, 817–822.
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075.
- Nunes, R.J.; de Oliveira, P.; Lages, A.; Becker, J.D.; Marcelino, P.; Barroso, E.; Perdigoto, R.; Kelly, J.W.; Quintas, A.; Santos, S.C.R. Transthyretin Proteins Regulate Angiogenesis by Conferring Different Molecular Identities to Endothelial Cells*. J. Biol. Chem. 2013, 288, 31752–31760.
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue Damage in the Amyloidoses: Transthyretin Monomers and Nonnative Oligomers Are the Major Cytotoxic Species in Tissue Culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822.
- Burton, G.J.; Yung, H.-W. Endoplasmic Reticulum Stress in the Pathogenesis of Early-Onset Pre-Eclampsia. Pregnancy Hypertens. 2011, 1, 72–78.
- Hemagirri, M.; Chen, Y.; Gopinath, S.C.B.; Sahreen, S.; Adnan, M.; Sasidharan, S. Crosstalk between Protein Misfolding and Endoplasmic Reticulum Stress during Ageing and Their Role in Age-Related Disorders. Biochimie 2023.
- Yoshida, H. ER Stress and Diseases. FEBS J. 2007, 274, 630–658.
- Cheng, S.; Huang, Z.; Banerjee, S.; Jash, S.; Buxbaum, J.N.; Sharma, S. Evidence from Human Placenta, Endoplasmic Reticulum–Stressed Trophoblasts, and Transgenic Mice Links Transthyretin Proteinopathy to Preeclampsia. Hypertension 2022, 79, 1738–1754.
- Du, L.; He, F.; Kuang, L.; Tang, W.; Li, Y.; Chen, D. eNOS/iNOS and Endoplasmic Reticulum Stress-Induced Apoptosis in the Placentas of Patients with Preeclampsia. J. Hum. Hypertens. 2017, 31, 49–55.
- Expression of Markers of Endoplasmic Reticulum Stress-Induced Apoptosis in the Placenta of Women with Early and Late Onset Severe Pre-Eclampsia—ClinicalKey. Available online: https://www.clinicalkey.com/#!/content/playContent/1-s2.0-S1028455914002022?returnurl=https:%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1028455914002022%3Fshowall%3Dtrue&referrer=https:%2F%2Fpubmed.ncbi.nlm.nih.gov%2F (accessed on 22 November 2023).
- Lian, I.A.; Løset, M.; Mundal, S.B.; Fenstad, M.H.; Johnson, M.P.; Eide, I.P.; Bjørge, L.; Freed, K.A.; Moses, E.K.; Austgulen, R. Increased Endoplasmic Reticulum Stress in Decidual Tissue from Pregnancies Complicated by Fetal Growth Restriction with and without Pre-Eclampsia. Placenta 2011, 32, 823–829.
- Yung, H.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of Placental Translation Inhibition and Endoplasmic Reticulum Stress in the Etiology of Human Intrauterine Growth Restriction. Am. J. Pathol. 2008, 173, 451–462.
- Mochan, S.; Dhingra, M.K.; Gupta, S.K.; Saxena, S.; Arora, P.; Yadav, V.; Rani, N.; Luthra, K.; Dwivedi, S.; Bhatla, N.; et al. Status of VEGF in Preeclampsia and Its Effect on Endoplasmic Reticulum Stress in Placental Trophoblast Cells. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2019, 4, 100070.
- Iyer, K.; Chand, K.; Mitra, A.; Trivedi, J.; Mitra, D. Diversity in Heat Shock Protein Families: Functional Implications in Virus Infection with a Comprehensive Insight of Their Role in the HIV-1 Life Cycle. Cell Stress. Chaperones 2021, 26, 743–768.
- Tripathi, A.; Iyer, K.; Mitra, D. HIV-1 Replication Requires Optimal Activation of the Unfolded Protein Response. FEBS Lett. 2023, 597, 2908–2930.
- Jee, B.; Dhar, R.; Singh, S.; Karmakar, S. Heat Shock Proteins and Their Role in Pregnancy: Redefining the Function of “Old Rum in a New Bottle”. Front. Cell Dev. Biol. 2021, 9, 648463.
- Rai, S.; Tapadia, M.G. Hsc70-4 Aggravates PolyQ-Mediated Neurodegeneration by Modulating NF-κB Mediated Immune Response in Drosophila. Front. Mol. Neurosci. 2022, 15, 857257.
- Cater, J.H.; Kumita, J.R.; Zeineddine Abdallah, R.; Zhao, G.; Bernardo-Gancedo, A.; Henry, A.; Winata, W.; Chi, M.; Grenyer, B.S.F.; Townsend, M.L.; et al. Human Pregnancy Zone Protein Stabilizes Misfolded Proteins Including Preeclampsia- and Alzheimer’s-Associated Amyloid Beta Peptide. Proc. Natl. Acad. Sci. USA 2019, 116, 6101–6110.
- Devriendt, K.; Van den Berghe, H.; Cassiman, J.-J.; Marynen, P. Primary Structure of Pregnancy Zone Protein. Molecular Cloning of a Full-Length PZP cDNA Clone by the Polymerase Chain Reaction. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1991, 1088, 95–103.
- Sottrup-Jensen, L.; Folkersen, J.; Kristensen, T.; Tack, B.F. Partial Primary Structure of Human Pregnancy Zone Protein: Extensive Sequence Homology with Human Alpha 2-Macroglobulin. Proc. Natl. Acad. Sci. USA 1984, 81, 7353–7357.
- Folkersen, J.; Teisner, B.; Grunnet, N.; Grudzinskas, J.G.; Westergaard, J.G.; Hindersson, P. Circulating Levels of Pregnancy Zone Protein: Normal Range and the Influence of Age and Gender. Clin. Chim. Acta 1981, 110, 139–145.
- Sand, O.; Folkersen, J.; Westergaard, J.G.; Sottrup-Jensen, L. Characterization of Human Pregnancy Zone Protein. Comparison with Human Alpha 2-Macroglobulin. J. Biol. Chem. 1985, 260, 15723–15735.
- Cater, J.H.; Mañucat-Tan, N.B.; Georgiou, D.K.; Zhao, G.; Buhimschi, I.A.; Wyatt, A.R.; Ranson, M. A Novel Role for Plasminogen Activator Inhibitor Type-2 as a Hypochlorite-Resistant Serine Protease Inhibitor and Holdase Chaperone. Cells 2022, 11, 1152.
- Wyatt, A.R.; Cater, J.H.; Ranson, M. PZP and PAI-2: Structurally-Diverse, Functionally Similar Pregnancy Proteins? Int. J. Biochem. Cell Biol. 2016, 79, 113–117.
- Ohkuchi, A.; Minakami, H.; Aoya, T.; Haga, T.; Kimura, H.; Suzuki, M.; Sato, I. Expansion of the Fraction of Th1 Cells in Women with Preeclampsia: Inverse Correlation between the Percentage of Th1 Cells and the Plasma Level of PAI-2. Am. J. Reprod. Immunol. 2001, 46, 252–259.
- Saito, S.; Shima, T.; Nakashima, A. Immunological Maladaptation. In Preeclampsia: Basic, Genomic, and Clinical; Saito, S., Ed.; Comprehensive Gynecology and Obstetrics; Springer: Singapore, 2018; pp. 65–84. ISBN 978-981-10-5891-2.
- Grancha, S.; Estellés, A.; Gilabert, J.; Chirivella, M.; España, F.; Aznar, J. Decreased Expression of PAI-2 mRNA and Protein in Pregnancies Complicated with Intrauterine Fetal Growth Retardation. Thromb. Haemost. 1996, 76, 761–767.
- Roes, E.M.; Sweep, C.G.F.; Thomas, C.M.; Zusterzeel, P.L.; Geurts-Moespot, A.; Peters, W.H.; Steegers, E.A. Levels of Plasminogen Activators and Their Inhibitors in Maternal and Umbilical Cord Plasma in Severe Preeclampsia. Am. J. Obstet. Gynecol. 2002, 187, 1019–1025.
- de Graff, A.M.; Mosedale, D.E.; Sharp, T.; Dill, K.A.; Grainger, D.J. Proteostasis Is Adaptive: Balancing Chaperone Holdases against Foldases. PLoS Comput. Biol. 2020, 16, e1008460.
- Lee, J.A.; Yerbury, J.J.; Farrawell, N.; Shearer, R.F.; Constantinescu, P.; Hatters, D.M.; Schroder, W.A.; Suhrbier, A.; Wilson, M.R.; Saunders, D.N.; et al. SerpinB2 (PAI-2) Modulates Proteostasis via Binding Misfolded Proteins and Promotion of Cytoprotective Inclusion Formation. PLoS ONE 2015, 10, e0130136.
- Nakashima, A.; Cheng, S.-B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y.; et al. Evidence for Lysosomal Biogenesis Proteome Defect and Impaired Autophagy in Preeclampsia. Autophagy 2020, 16, 1771–1785.
- Cheng, S.-B.; Nakashima, A.; Sharma, S. Understanding Pre-Eclampsia Using Alzheimer’s Etiology: An Intriguing Viewpoint. Am. J. Reprod. Immunol. 2016, 75, 372–381.
- Nakashima, A.; Cheng, S.-B.; Kusabiraki, T.; Motomura, K.; Aoki, A.; Ushijima, A.; Ono, Y.; Tsuda, S.; Shima, T.; Yoshino, O.; et al. Endoplasmic Reticulum Stress Disrupts Lysosomal Homeostasis and Induces Blockade of Autophagic Flux in Human Trophoblasts. Sci. Rep. 2019, 9, 11466.
- Nakashima, A.; Tsuda, S.; Kusabiraki, T.; Aoki, A.; Ushijima, A.; Shima, T.; Cheng, S.-B.; Sharma, S.; Saito, S. Current Understanding of Autophagy in Pregnancy. Int. J. Mol. Sci. 2019, 20, 2342.
- Nakashima, A.; Shima, T.; Tsuda, S.; Aoki, A.; Kawaguchi, M.; Furuta, A.; Yasuda, I.; Yoneda, S.; Yamaki-Ushijima, A.; Cheng, S.-B.; et al. Aggrephagy Deficiency in the Placenta: A New Pathogenesis of Preeclampsia. Int. J. Mol. Sci. 2021, 22, 2432.
- Nakashima, A.; Yamanaka-Tatematsu, M.; Fujita, N.; Koizumi, K.; Shima, T.; Yoshida, T.; Nikaido, T.; Okamoto, A.; Yoshimori, T.; Saito, S. Impaired Autophagy by Soluble Endoglin, under Physiological Hypoxia in Early Pregnant Period, Is Involved in Poor Placentation in Preeclampsia. Autophagy 2013, 9, 303–316.
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal 2014, 20, 460–473.
- Opoku-Nsiah, K.A.; Gestwicki, J.E. Aim for the Core: Suitability of the Ubiquitin-Independent 20S Proteasome as a Drug Target in Neurodegeneration. Transl. Res. 2018, 198, 48–57.
- Nakashima, A.; Shima, T.; Aoki, A.; Kawaguchi, M.; Yasuda, I.; Tsuda, S.; Yoneda, S.; Yamaki-Ushijima, A.; Cheng, S.; Sharma, S.; et al. Placental Autophagy Failure: A Risk Factor for Preeclampsia. J. Obstet. Gynaecol. Res. 2020, 46, 2497–2504.
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an Autophagy Inhibitor, Potentiates the Radiosensitivity of Glioma Initiating Cells by Inhibiting Autophagy and Activating Apoptosis. BMC Neurol. 2016, 16, 178.
- Owaraganise, A.; Migisha, R.; Ssalongo, W.G.M.; Tibaijuka, L.; Kayondo, M.; Twesigomwe, G.; Ngonzi, J.; Lugobe, H.M. Nonproteinuric Preeclampsia among Women with Hypertensive Disorders of Pregnancy at a Referral Hospital in Southwestern Uganda. Obstet. Gynecol. Int. 2021, 2021, 9751775.
- Palmqvist, S.; Insel, P.S.; Stomrud, E.; Janelidze, S.; Zetterberg, H.; Brix, B.; Eichenlaub, U.; Dage, J.L.; Chai, X.; Blennow, K.; et al. Cerebrospinal Fluid and Plasma Biomarker Trajectories with Increasing Amyloid Deposition in Alzheimer’s Disease. EMBO Mol. Med. 2019, 11, e11170.
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-Precision Plasma β-Amyloid 42/40 Predicts Current and Future Brain Amyloidosis. Neurology 2019, 93, e1647–e1659.
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High Performance Plasma Amyloid-β Biomarkers for Alzheimer’s Disease. Nature 2018, 554, 249–254.
- Stevenson-Hoare, J.; Heslegrave, A.; Leonenko, G.; Fathalla, D.; Bellou, E.; Luckcuck, L.; Marshall, R.; Sims, R.; Morgan, B.P.; Hardy, J.; et al. Plasma Biomarkers and Genetics in the Diagnosis and Prediction of Alzheimer’s Disease. Brain 2023, 146, 690–699.
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; Hansson, O. Prediction of Future Alzheimer’s Disease Dementia Using Plasma Phospho-Tau Combined with Other Accessible Measures. Nat. Med. 2021, 27, 1034–1042.
- McCarthy, F.P.; Adetoba, A.; Gill, C.; Bramham, K.; Bertolaccini, M.; Burton, G.J.; Girardi, G.; Seed, P.T.; Poston, L.; Chappell, L.C. Urinary Congophilia in Women with Hypertensive Disorders of Pregnancy and Preexisting Proteinuria or Hypertension. Am. J. Obstet. Gynecol. 2016, 215, 464.e1–464.e7.
- Nagarajappa, C.; Rangappa, S.S.; Suryanarayana, R.; Balakrishna, S. Urinary Congophilia in Preeclampsia: Experience from a Rural Tertiary-Care Hospital in India. Pregnancy Hypertens. 2018, 13, 83–86.
- Khaliq, O.P.; Phoswa, W.N.; Moodley, J. The Effectiveness of the Congo Red Dot Paper Test in Hypertensive Disorders of Pregnancy: A Systematic Review and Meta-Analysis. Front. Reprod. Health 2023, 5, 1120937.
- Wong, N.K.L.; Wah, I.Y.M.; Wong, S.T.K.; Nguyen-Hoang, L.; Lau, C.S.L.; Ip, P.N.P.; Leung, H.H.Y.; Sahota, D.S.; Poon, L.C. A Point-of-Care Urine Test to Predict Adverse Maternal and Neonatal Outcomes in Asian Women with Suspected Preeclampsia. Arch. Gynecol. Obstet. 2023, 1–10.
- Wong, S.T.K.; Sahota, D.S.; Wong, N.K.L.; Wah, I.Y.M.; Wang, X.; Lau, S.L.; Chiu, C.P.H.; Ip, P.N.P.; Poon, L.C. A Point-of Care Urine Test to Predict Preeclampsia Development in Asian Women with Suspected Preeclampsia. Pregnancy Hypertens. 2023, 32, 28–34.
- Sailakshmi, M.P.A.; Prabhu, M.R.; Prabhakara, S.; Anbazhagan, K.; Rupakala, B.M. Congo Red Dot Test in the Early Prediction and Diagnosis of Pre-Eclampsia in a Tertiary Health Care Centre in India. Pregnancy Hypertens. 2021, 25, 225–229.
- Sammar, M.; Syngelaki, A.; Sharabi-Nov, A.; Nicolaides, K.; Meiri, H. Can Staining of Damaged Proteins in Urine Effectively Predict Preeclampsia? Fetal Diagn. Ther. 2017, 41, 23–31.
- Younis, D.; Mosbah, A.; Zakaria, M.M.; Awadalla, A.; El-Kannishy, G.; Shemies, R.S. Urinary Congophilia in Pregnancy: A Marker of Kidney Injury Rather than Preeclampsia. J. Hypertens. 2023, 41, 1760–1767.
- Döbert, M.; Varouxaki, A.-N.; Mu, A.C.; Syngelaki, A.; Nicolaides, K.H. Screening for Late Preeclampsia at 35-37 Weeks by the Urinary Congo-Red Dot Paper Test. J. Matern. Fetal Neonatal Med. 2022, 35, 5686–5690.
- Li, X.-M.; Liu, X.-M.; Xu, J.; Du, J.; Cuckle, H. Late Pregnancy Screening for Preeclampsia with a Urinary Point-of-Care Test for Misfolded Proteins. PLoS ONE 2020, 15, e0233214.
- Bracken, H.; Buhimschi, I.A.; Rahman, A.; Smith, P.R.S.; Pervin, J.; Rouf, S.; Bousieguez, M.; López, L.G.; Buhimschi, C.S.; Easterling, T.; et al. Congo Red Test for Identification of Preeclampsia: Results of a Prospective Diagnostic Case-Control Study in Bangladesh and Mexico. EClinicalMedicine 2021, 31, 100678.
- Rood, K.M.; Buhimschi, C.S.; Dible, T.; Webster, S.; Zhao, G.; Samuels, P.; Buhimschi, I.A. Congo Red Dot Paper Test for Antenatal Triage and Rapid Identification of Preeclampsia. EClinicalMedicine 2019, 8, 47–56.
- Millen, K.R.; Buhimschi, C.S.; Zhao, G.; Rood, K.M.; Tabbah, S.; Buhimschi, I.A. Serum and urine thioflavin T (ThT) enhanced fluorescence in severe preeclampsia. Hypertension 2018, 71, 1185–1192.
- Rodriguez Chavez, J.L.; Fuentes Gutiérrez, E.K.; de Jesús Angeles Vázquez, M.; Mendieta Zerón, H. Evaluation of Congo Red Staining Kit to Determine Proteinuria in Preeclampsia. J. Clin. Diagn. Res. 2018, 12, QM01–QM05.
- Planté-Bordeneuve, V.; Said, G. Familial Amyloid Polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097.
- Cardoso, I.; Goldsbury, C.S.; Müller, S.A.; Olivieri, V.; Wirtz, S.; Damas, A.M.; Aebi, U.; Saraiva, M.J. Transthyretin Fibrillogenesis Entails the Assembly of Monomers: A Molecular Model for in Vitro Assembled Transthyretin Amyloid-like Fibrils. J. Mol. Biol. 2002, 317, 683–695.
- Cotrina, E.Y.; Santos, L.M.; Rivas, J.; Blasi, D.; Leite, J.P.; Liz, M.A.; Busquets, M.A.; Planas, A.; Prohens, R.; Gimeno, A.; et al. Targeting Transthyretin in Alzheimer’s Disease: Drug Discovery of Small-Molecule Chaperones as Disease-Modifying Drug Candidates for Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 226, 113847.
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER Stress and UPR in Alzheimer’s Disease: Mechanisms, Pathogenesis, Treatments. Cell Death Dis. 2022, 13, 706.
- Huang, L.-K.; Kuan, Y.-C.; Lin, H.-W.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease: A 2020–2023 Update. J. Biomed. Sci. 2023, 30, 83.
- Ji, C.; Sigurdsson, E.M. Current Status of Clinical Trials on Tau Immunotherapies. Drugs 2021, 81, 1135–1152.
- Eshraghi, M.; Ahmadi, M.; Afshar, S.; Lorzadeh, S.; Adlimoghaddam, A.; Rezvani Jalal, N.; West, R.; Dastghaib, S.; Igder, S.; Torshizi, S.R.N.; et al. Enhancing Autophagy in Alzheimer’s Disease through Drug Repositioning. Pharmacol. Ther. 2022, 237, 108171.
- Fang, J.; Zhang, P.; Zhou, Y.; Chiang, C.-W.; Tan, J.; Hou, Y.; Stauffer, S.; Li, L.; Pieper, A.A.; Cummings, J.; et al. Endophenotype-Based in Silico Network Medicine Discovery Combined with Insurance Record Data Mining Identifies Sildenafil as a Candidate Drug for Alzheimer’s Disease. Nat. Aging 2021, 1, 1175–1188.
- El-Bakly, W.; Wagdy, O.; Sobhy, A.; Abo Elenain, O.; Riad, M.S.; El Sayed, M.; Tarkhan, S.; Yassen, M.; Mahmoud, A.; Bassiony, M.; et al. The Efficacy and Underlying Mechanism of Phosphodiesterase- 5 Inhibitors in Preventing Cognitive Impairment and Alzheimer Pathology: A Systematic Review of Animal Studies. Behav. Brain Res. 2019, 372, 112004.
- Desai, R.J.; Mahesri, M.; Lee, S.B.; Varma, V.R.; Loeffler, T.; Schilcher, I.; Gerhard, T.; Segal, J.B.; Ritchey, M.E.; Horton, D.B.; et al. No Association between Initiation of Phosphodiesterase-5 Inhibitors and Risk of Incident Alzheimer’s Disease and Related Dementia: Results from the Drug Repurposing for Effective Alzheimer’s Medicines Study. Brain Commun. 2022, 4, fcac247.
- Andraweera, P.H.; Lassi, Z.S. Cardiovascular Risk Factors in Offspring of Preeclamptic Pregnancies-Systematic Review and Meta-Analysis. J. Pediatr. 2019, 208, 104–113.e6.
- Nahum Sacks, K.; Friger, M.; Shoham-Vardi, I.; Spiegel, E.; Sergienko, R.; Landau, D.; Sheiner, E. Prenatal Exposure to Preeclampsia as an Independent Risk Factor for Long-Term Cardiovascular Morbidity of the Offspring. Pregnancy Hypertens. 2018, 13, 181–186.
- Ray, J.G.; Vermeulen, M.J.; Schull, M.J.; Redelmeier, D.A. Cardiovascular Health after Maternal Placental Syndromes (CHAMPS): Population-Based Retrospective Cohort Study. Lancet 2005, 366, 1797–1803.
- Wu, P.; Haththotuwa, R.; Kwok, C.S.; Babu, A.; Kotronias, R.A.; Rushton, C.; Zaman, A.; Fryer, A.A.; Kadam, U.; Chew-Graham, C.A.; et al. Preeclampsia and Future Cardiovascular Health. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003497.
- Andolf, E.; Bladh, M.; Möller, L.; Sydsjö, G. Prior Placental Bed Disorders and Later Dementia: A Retrospective Swedish Register-Based Cohort Study. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 1090–1099.
- Basit, S.; Wohlfahrt, J.; Boyd, H.A. Pre-Eclampsia and Risk of Dementia Later in Life: Nationwide Cohort Study. BMJ 2018, 363, k4109.

