Protein misfolding disorders are a group of diseases characterized by supra-physiologic accumulation and aggregation of pathogenic proteoforms resulting from improper protein folding and/or insufficiency in clearance mechanisms. Although these processes have been historically linked to neurodegenerative disorders, such as Alzheimer’s disease, evidence linking protein misfolding to other pathologies continues to emerge. Indeed, the deposition of toxic protein aggregates in the form of oligomers or large amyloid fibrils has been linked to type 2 diabetes, various types of cancer, and, in more recent years, to preeclampsia, a life-threatening pregnancy-specific disorder. While extensive physiological mechanisms are in place to maintain proteostasis, processes, such as aging, genetic factors, or environmental stress in the form of hypoxia, nutrient deprivation or xenobiotic exposures can induce failure in these systems. As such, pregnancy, a natural physical state that already places the maternal body under significant physiological stress, creates an environment with a lower threshold for aberrant aggregation.
1. Introduction
Proteostasis, which describes the process through which protein homeostasis is maintained in the body, is achieved through a collection of tightly regulated mechanisms and systems. These systems include regulation of protein synthesis at translational and post-translational levels, folding into structurally functional entities through a network of molecular chaperones, and degradation through autophagy or the ubiquitin–proteasome pathway [
1]. Although these systems are tightly regulated, they must also be dynamic to adapt to fluctuating physiological conditions. Unfortunately, there are multitudes of conditions that are known to disrupt protein homeostasis, leading to aberrant polymerization and aggregation of inert or toxic higher-order protein species in tissues. It was even postulated that protein misfolding pathology could be an underlying factor in half of all human diseases [
2,
3]. Some of the well-studied disorders in which loss of proteostasis is known to contribute to pathogenesis include cystic fibrosis, diabetes, α-antitrypsin deficiency, cancer, and neurodegenerative disorders, like Alzheimer’s disease [
3]. Protein misfolding is also known to be precipitated by aging or major physiological alterations and stress [
4]. One such stress that involves drastic anatomic, metabolic, and cardiovascular changes is pregnancy.
Pregnancy has been described by Williams and David as a maternal stress test [
5]. Indeed, pregnancy puts women through a series of rapid physiological changes that are necessary to accommodate the demands of gestation. Along with increased cardiac output and basal metabolic rate, increased glucose synthesis, increased insulin resistance, and hyperlipidemia, there is increased protein synthesis and turnover to accommodate the higher rate of tissue synthesis [
6,
7,
8,
9,
10].
2. The Central Hypothesis of PE Pathophysiology and Current Gaps
While substantial progress has been made over the years to understand the pathophysiological mechanisms associated with PE, significant controversy remains. A chief challenge is that PE is not a single disease with a single causative pathway but rather a collection of highly heterogeneous clinical phenotypes with non-specific molecular and pathologic findings that often overlap with other pregnancy disorders [
17,
18]. Clinically, PE manifests as new-onset hypertension (systolic blood pressure ≥ 140 mmHg, diastolic blood pressure ≥ 90 mmHg) after 20 weeks of gestation with or without proteinuria [
17]. This definition is a recent revision of the traditional clinical classification criteria for PE, which was based on the combination of hypertension and proteinuria [
19,
20]. The definition has been broadened to encompass cases without proteinuria but with evidence of maternal end-organ damage in the form of hemolysis, elevated liver enzymes and low platelet count syndrome (HELLP); renal damage; pulmonary edema; visual disturbances; persistent headache; stroke; and seizures (eclampsia) [
19]. The current diagnostic and clinical guidelines distinguish between PE with and without severe features (sPE) based on the occurrence of any of these severe maternal symptoms or a systolic blood pressure over 160 mmHg and diastolic blood pressure above 90 mmHg [
19]. As a result of this revision, an increased number of women with milder symptoms and a lower probability of adverse outcomes are now diagnosed with PE [
21]. On the other hand, the clinical phenotype can also be subclassified based on the time of disease onset. PE arising before 34 weeks of gestation classifies as early-onset PE (EOPE) while PE arising after 34 weeks of gestation classifies as late-onset PE (LOPE). LOPE and EOPE have been shown to have distinct pathophysiological mechanisms and varying levels of disease severity [
17].
It is widely accepted that the placenta, a transient but indispensable organ for pregnancy maintenance and fetal development, is the initial site where dysfunction arises. Indeed, the central hypothesis of PE pathogenesis supports a two-stage model with a defective invasion of extravillous trophoblasts cells (EVTs) and incomplete remodeling of spiral arteries at the maternal–fetal interface during placentation as the inciting factor for disease (Stage 1) [
23,
24,
25]. In normal pregnancy, spiral arteries, which connect the placenta to the maternal blood supply, undergo extensive transformation. These blood vessels must widen to accommodate the increase in blood flow necessary to support a growing fetus [
26]. This remodeling process has been shown to be mediated by the invasion of EVTs from the anchoring villi to the maternal decidua and deeper into the myometrium (interstitial trophoblasts). A subpopulation of the migrating EVTs also invades the wall and lumen of spiral arteries, where they replace endothelial cells (as intramural and endovascular trophoblasts, respectively) [
26,
27,
28,
29,
30].
Despite well-established evidence of this pathophysiologic process occurring in PE, the shallow trophoblast invasion characterizing Stage 1 is not unique to PE. The failure of spiral artery transformation has also been described in preterm birth, fetal growth restriction (FGR), placental accreta spectrum disorders, and spontaneous miscarriage with varying degrees of changes and severity in placental phenotype [
32,
34,
36,
37,
38,
39]. Furthermore, although signs of abnormal placentation are commonly observed, they are not observed in all affected women, nor are they always sufficient to trigger clinical symptoms when present [
29,
40]. Intriguingly, the endothelial dysfunction resulting from placental malperfusion causes an imbalance in angiogenic markers, such as soluble fms-like tyrosine kinase-1 (sFlt-1) andsoluble endoglin (sEng), which increase; and VEGF which decreases [
41,
42,
43]. However, there is evidence that these changes in levels of angiogenic factors can be detected as early as 7 weeks of gestation, before the deep invasion of EVTs occurs, which suggests that vascular remodeling dysfunction may occur independently of trophoblast invasion [
17,
33,
44,
45].
A multitude of environmental, genetic, fetal, and maternal risk factors that modulate PE risk and influence disease severity also exist. One striking example is the relationship between chronic kidney disease (CKD) and PE. On one hand, PE onset is associated with the development of acute end-organ damage involving vascular damage, glomerular endotheliosis, and chronic kidney damage, with a three–fifteen-fold increased relative risk of subsequent end-stage renal disease [
53,
54]. On the other hand, women who have pre-existing CKD are ten times more likely to develop PE compared to women without CKD [
55,
56]. Even more intriguing, women with a single kidney are three times more likely to develop PE than women with two kidneys, even in the setting of a normal glomerular filtration rate [
57]. This epidemiological observation has also been demonstrated in vivo in a mouse model that exhibits a PE-like phenotype following unilateral nephrectomy [
58].
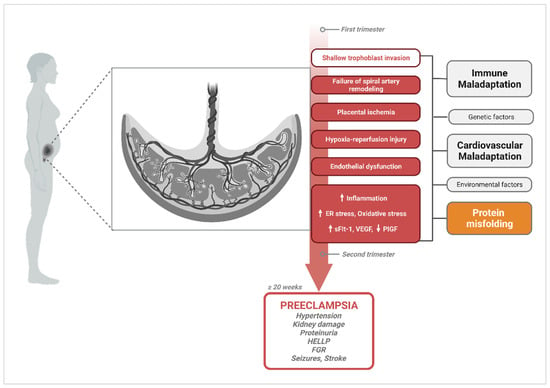
The spectrum of potential placental, fetal, and maternal pathophysiological processes and risk factors involved in PE create considerable ambiguity in discriminating cause from effect. Hence, a multifactorial etiology of PE that involves abnormal placentation, immune maladaptation mechanisms, cardiovascular and renal dysfunction, as well as multisystemic disruptions in proteostasis cannot be dismissed (Figure 1).
Figure 1. Diagram of pathophysiological processes described in the central hypothesis of PE pathogenesis, illustrating multifactorial etiologies that could trigger the widely accepted canonical pathway. Canonical mechanisms involved in PE pathogenesis and their progression to PE symptomatology are highlighted in red. Upward arrows indicate the pathological processes and vascular markers that increase in PE, while downward arrows represent markers that are known to decrease. A handful of other alternate or complementary etiological factors that have been proposed to contribute to these central mechanisms are shown in grey (immune maladaptation, genetic factors, cardiovascular maladaptation, environmental factors); and orange for protein misfolding. Created with
BioRender.com, accessed on 18 December 2023.
3. Protein Misfolding in PE
3.1. General Protein Misfolding and Aggregation Mechanisms
Protein misfolding mechanisms and the protein conformational disorders (proteinopathies) that arise as a result have been extensively studied in the context of neurodegenerative disorders, such as AD, Parkinson’s, Huntington’s, and prion disease. More and more illnesses continue to be characterized as proteinopathies thanks to the advances made while studying these prototypical protein conformational disorders. It is well recognized that protein molecules can assume versatile secondary, tertiary, and quaternary structures or conformations based on their function and biological environment [
1,
59]. Polypeptide chains derived following mRNA translation must go through folding and assembly into well-defined native structures to assume the functional state of the protein. These processes occur primarily in the endoplasmic reticulum (ER), a subcellular compartment that serves many functions, including the acquisition of post-translational modifications, but can also take place in the cytoplasm [
1,
60]. Dysregulation of normal folding processes at this level, leading to improper folding and assembly into non-native proteoforms, can occur due to the failure of molecular chaperones, ER-associated protein complexes that facilitate proper protein folding, or due to disturbances in the physiologic environment that can trigger the abnormal polymerization of peptides [
1,
61]. Accumulation of abnormal protein conformations can also result from failure in protein degradation pathways, such as disrupted ubiquitin–proteasome signaling or ineffective cellular autophagy processes [
62,
63].
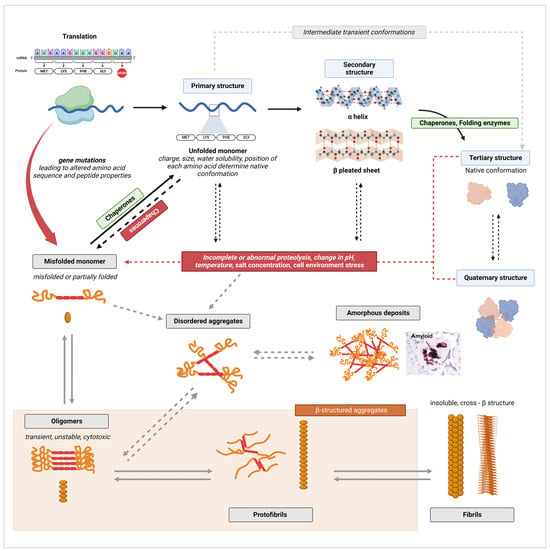
The pathways of native protein structure formation and of amyloid fibril formation are contrasted in
Figure 2. As established by decades of research in biochemistry and protein biology, the amino acid sequence of a peptide determines the unique functional conformation a protein will adopt [
64,
65]. Each unique protein sequence and each amino acid in the sequence carries intrinsic properties, such as size, charge, or water solubility (hydrophobicity or hydrophily), that drive the interatomic interactions responsible for shaping a protein into its optimal conformation [
66]. A normal physiological environment is also fundamental for proper folding as native conformations must be thermodynamically stable under those conditions [
65,
66]. In ideal physiological conditions (which vary for each protein), an unfolded monomeric peptide (primary structure) will assume multiple potential conformational states (transient intermediates) that are accessible to the peptide until the most energetically favorable conformation is attained [
59,
65,
66,
67]. These intermediates include partially folded secondary structures, such as α helixes and β-pleated sheet structures, which form primarily through non-covalent interactions. These non-covalent bonds include electrostatic forces (ionic bonds); van der Waals interactions; and, most critically, intramolecular (both α helixes and β-sheet) and intermolecular hydrogen bonds (β-sheet only) [
68,
69,
70].
Figure 2. Illustrative diagram of native protein structure formation and amyloid fibril formation pathways. Canonical processes are indicated by full arrows, while alternate pathways are indicated by dashed arrows. Black arrows were used for processes of normal protein folding, while grey arrows were used to indicate fibrillation mechanisms. Created with
BioRender.com, accessed on 20 January 2024.
On the other hand, disturbances in the cellular environment, such as changes in pH, temperature, salt concentration, or aberrant proteolysis, can lead to the formation of misfolded peptides [
72]. The aforementioned disturbances can cause the disruption of native bonds and aberrant non-covalent interactions, leading to misfolding (
Figure 2). The thermodynamic stability of a protein’s native conformation can also be disrupted by protein concentrations that exceed a specific critical threshold [
73]. In such conditions, the formation of aggregates becomes energetically more favorable than maintaining the native state [
67,
73].
The first stage in the aggregation of unfolded, partially folded, or misfolded monomers is the formation of oligomers [
66]. Oligomers are unstable polymers of at least four repeating units of a monomer, connected through weak and non-specific intermolecular interactions (
Figure 2) [
59,
66]. As monomers continue to be added by self-association, oligomers transform from disorganized amorphous aggregates into more compact, structured, and stable aggregates known as protofilaments, which further self-assemble into protofibrils [
59,
66,
77]. Two to eight protofibrils then intertwine on a lateral axis to form mature fibrils with β-strands running perpendicular to the fibril axis [
59]. Extensive hydrogen bonding within and between β-sheets plays a critical role in stabilizing the highly ordered cross-β structure [
72]. This self-assembly process occurs through nucleated polymerization [
67]. The classical mechanism of nucleation is described as primary nucleation, in which the rapid self-propagation of monomers into larger fibrils is initiated once a nuclei, the smallest oligomeric species that can trigger self-growth and elongation, is formed [
78,
79,
80,
81].
In AD and other protein misfolding disorders, the presence of fibril deposits in tissue was thought to be the main pathogenic driver of disease. However, strong evidence supporting oligomers as the most toxic and pathogenic conformation has now emerged [
86]. Oligomers have been shown to be highly cytotoxic to neuronal cells and to disrupt cell membrane integrity, calcium homeostasis, and essential cellular processes and signaling pathways [
86,
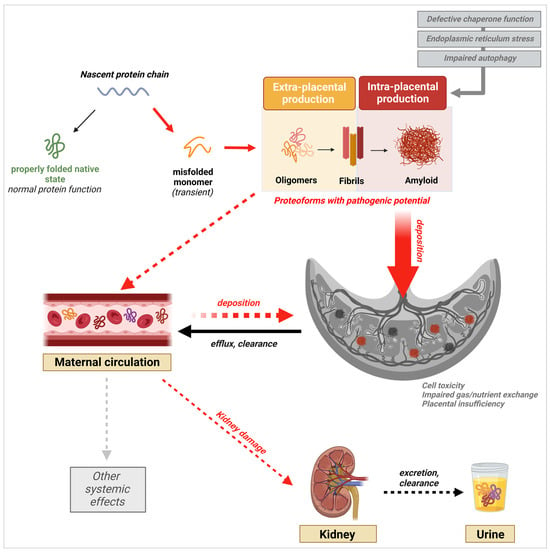
87]. This knowledge has triggered a similar shift in our understanding of other proteinopathies, including PE. Indeed, the latest research findings demonstrate the presence and potential pathogenicity of prefibrillar species of diverse proteins in PE. Nevertheless, the evidence also shows that fibrils and large amyloid plaques are present in the placenta, as they are in the brain of AD patients, supporting a role for both species in PE pathophysiology, albeit through different mechanisms (
Figure 3) [
87].
Figure 3. Schematic of the protein aggregation cascade and their potential multisystemic effects leading to or resulting from PE onset. Validated processes are shown by full arrows, while proposed potential mechanisms are indicated by dashed arrows. Created with
BioRender.com, accessed on 20 January 2024.
3.2. Excretion of Misfolded Proteins in the Urine
Initial clues that protein misfolding pathology occurs in PE came to light when a group identified the presence of supramolecular aggregates of SERPINA1 (alpha-1 antitrypsin, A1AT) in the urine of women with sPE, along with conformational aggregates (including oligomers) of the protein in the placenta [
13]. Since proteinuria is a major clinical manifestation of PE, scholars further verified the presence of misfolded proteins in the urine of women with sPE compared to gestational age-matched controls using Congo red, a dye that exhibits high binding affinity for amyloid proteins with a β-sheet conformation and is the gold standard for post-mortem amyloid plaque detection in AD [
12,
89].
3.3. Presence of Misfolded Proteins in the Maternal Circulation
Kalkunte et al. demonstrated the presence of aggregated proteoforms of transthyretin (TTR) in the serum of women with PE, providing additional supporting evidence for the protein misfolding hypothesis of PE pathophysiology [
11]. They also showed that the intraperitoneal injection of PE serum or TTR directly extracted from PE serum induced a PE-like syndrome characterized by hypertension, glomerular endotheliosis, fetal growth restriction, proteinuria, and increased sFlt-1 and sEng in IL-10
-/- mice compared to mice injected with serum from normal pregnancies [
11]. The PE-like phenotype was rescued when PE serum was co-administered with exogenous TTR in its native form or when TTR was depleted from PE serum [
11]. These findings highly suggest that the presence of these abnormal TTR aggregates is causative of PE and not a result of any preceding dysfunction. Using a cell-based in vitro model for the detection of protein aggregates, Cheng et al. also reported the presence of Amyloid-β in addition to TTR in the serum of women with EOPE and LOPE [
15]. Using the same technique, Jash et al. confirmed that aggregated forms of the cis stereo-isoform of P-tau231 (phosphorylated at threonine residue 231), a protein isoform linked to pathogenicity in AD, are present in the serum of women with EOPE and LOPE but not the gestational-age-matched controls. The presence of multiple proteins that have been proven to be pathogenic in AD in PE reinforces the idea that the aggregation and accumulation of these proteoforms in PE serum is not harmless.
3.4. Placental Deposition of Extracellular Protein Aggregates
A group has previously reported the presence of amyloid-like deposits of SERPINA1, APP, and Aβ in the placentas of women with sPE [
12,
13]. Detection of SERPINA1 aggregates using ATZ11, an antibody specific for oligomeric conformations of the protein, revealed increased placental deposition of misfolded SERPINA1 complexes in the stroma, endothelium, and fetal blood vessels of placentas from sPE pregnancies compared to gestational-age-matched controls [
13]. Furthermore, these deposits were predominantly localized in the endothelium. Given the central role endothelial damage plays in PE pathophysiology, and the well-documented toxicity of oligomeric protein conformations, the perivascular localization of these deposits could be an indication that they contribute to endothelial dysfunction [
86,
95].
Abnormal proteolytic cleavage of APP has been established as the mechanism for Aβ formation, aggregation, and accumulation in the AD brain [
59,
87,
96]. Similarly, hyperphosphorylation of tau, leading to the aberrant accumulation of pathogenic conformations of the protein, occurs [
96,
97]. Together, the deposition of toxic oligomers and large extracellular fibrillary plaques of Aβ with neurofibrillary tangles made of hyperphosphorylated tau are the two principal hallmarks of AD pathology. Along with the detection of misfolded SERPINA1, a group identified the presence of APP aggregates in the urine and placentas of women with sPE. Immunostaining of the placental tissue with ALZ90, an antibody specific for the Aβ fragment of APP, showed a positive signal for amyloid-like plaques of Aβ within areas of fibrinoid deposition [
12]. This phenotype was accompanied by significant upregulation of β-secretases, the enzymes that are responsible for the abnormal cleavage of APP into Aβ, in the trophoblast layer of placental villi. These observations led us to propose that the placental or systemic clearance of misfolded proteins might be deficient in PE, leading to a supraphysiologic load of these species in the urine.
Following the detection of aggregated proteoforms of TTR in PE serum, extensive studies have been conducted to understand their role in PE-associated placental pathology. Studies by Kalkunte et al. and Tong et al. revealed TTR deposition in the villous stroma and extravillous trophoblast cell layer in the human placental villi section [
11,
99]. The regions with positive TTR staining matched Thioflavin-S-positive regions, confirming that the TTR placental deposits in question were aggregated forms of the protein [
11]. Previous work by Kalkunte et al. demonstrated that PE serum containing TTR aggregates also caused the disruption of endovascular activity and crosstalk between endothelial cells and trophoblasts.
Although these latest research findings shed light on the placental metabolism of aggregated proteins, like Aβ, cis P-tau, and TTR, and offer a potential source for the aggregated proteoforms detected in serum, the conditions in which specific proteins are formed are still unclear. It also appears that pathogenic forms from different proteins may impact placental function at the same time, with some being more predominant than others. A deeper look into the function and characteristics of each of these proteins may help explain their presence and effects in PE.
4. PE-Associated Misfoldome
4.1. SERPINA1/α-1-Antitrypsin (A1AT)
Serine protease inhibitors, also known as serpins, are a superfamily of proteins involved in the regulation of proteolytic enzymes throughout the body [
100]. Serpins, especially SERPINA1, have been involved in a multitude of pathogenic processes, leading to a group of disorders recognized as serpinopathies. Notably, genetic mutations in SERPINA1 lead to severe and uncontrolled lung damage due to failure in the inhibition of neutrophil elastase, an abundant protease in the lung. In the liver, the main site of the synthesis of SERPINA1, defective SERPINA1 molecules accumulate and aggregate into higher-order polymers that induce fibrosis and cirrhosis [
101].
SERPINA1 is also an acute-phase reactant that is upregulated during inflammation, infection, and pregnancy [
101]. Thus, a role for the protein in PE pathophysiology is not unlikely. For example, the protein has well-documented anti-inflammatory properties that are independent of its proteolytic function [
108]. The increased placental and systemic inflammation, which are characteristic pathophysiological processes in PE, could potentially drive increased SERPINA1 expression as a protective mechanism [
92,
109]. On the other hand, Twina et al. showed that sPE is associated with lower circulating levels of SERPINA1, in which case dysregulation in SERPINA1 levels could be the cause for increased inflammation [
110]. It has also been proposed that SERPINA1 protects against the development of PE through the suppression of oxidative stress both in vitro and in vivo [
111,
112].
An important question is whether any of the known mutations that induce SERPINA1 oligomerization and deficiency exist in PE. Nagarajappa et al. conducted a cross-sectional study of 200 preeclamptic women to characterize the frequency of PiS and PiZ mutations, the two dominant mutations in the SERPINA1 gene that cause A1AT deficiency [
116]. Results showed that these two variants were non-existent in the study cohort; however, the presence of other unidentified pathogenic mutations cannot be excluded [
117]. The origin of SERPINA1 dysregulation and misfolding in PE is still unclear. Supposing the dysregulation results from established pathology as a compensatory mechanism instead of triggering it, there is still a high potential for a double-whammy effect due to the protein’s propensity to aggregate into toxic oligomers. The presence of highly amyloidogenic SERPINA1 fragments, such as the FVFLM peptide in the urine of women with PE, supports that excessive synthesis and release of toxic SERPINA1 proteoforms in the circulation either by the placenta or other organs occurs. The resulting urinary excretion of these peptides could be a clearance mechanism that helps remove these toxic species from the body [
13,
93].
4.2. Aβ and Its Precursor APP
Despite ample evidence of the presence of misfolded Aβ and APP in PE serum, placenta, and urine, mechanistic studies on their effects in PE are sparse [
12,
15,
94]. However, the aggregation of Aβ, which is a pathognomonic feature of AD pathology, is an uncanny similarity that can be exploited to better understand the mechanisms at play in PE. Thus, based on knowledge gathered from studies of Aβ pathology in AD, it can make inferences on what processes involving Aβ aggregation might be at play in PE. In AD, the amyloid cascade hypothesis is the classic view of pathogenesis [
96,
119]. According to this hypothesis, the deposition of Aβ aggregates drives neurotoxicity and the onset of dementia in AD [
119]. Aβ monomers are a cleavage product resulting from the non-canonical processing of APP by β- and γ- secretases enzymes (BACE1, BACE2, PS1, PS2) instead of α-secretases (ADAM10) [
120,
121].
4.3. Tau
Alongside Aβ, tau aggregation is a major driver and hallmark of AD symptomatology. Tau is a microtubule-associated protein that aids in the assembly and stabilization of microtubule filaments. In the nervous system, these functions are critical for neuronal maturation and axonal transport [
124]. Critically, the protein’s function is regulated via multiple post-translational modifications, the most common of which is phosphorylation at various residues [
125]. Phosphorylation of tau induces its dissociation from microtubules, causing alterations in cytoarchitecture and poor microtubule stability [
124,
125]. In AD, and other tau-associated proteinopathies, hyperphosphorylation of tau at specific sites is known to induce neurotoxic effects, including self-aggregation into pathogenic helical filaments that make up neurofibrillary tangles and toxic intracellular inclusions [
125].
4.4. TTR
TTR is an essential carrier protein that transports thyroxine and retinol-binding protein (RBP) bound to retinol (Vitamin A) [
126]. It is primarily synthesized by the liver, choroid plexus, and retina before being secreted into the circulation and cerebrospinal fluid (CSF) [
126]. Studies have shown that placental villous trophoblasts also synthesize TTR and that, in PE, an increased amount of placental extracellular vesicles carrying aggregated TTR as a cargo were released into maternal circulation [
99,
127].
One essential function of TTR that could also contribute to PE pathophysiology is its role in angiogenesis and maintaining blood–brain barrier integrity in AD [
129,
130]. Nunes et al. showed that V30M, an amyloidogenic hereditary variant of TTR, causes the downregulation of pro-angiogenic factor expression, induces apoptosis, and reduces cell migration in endothelial cells [
130]. Analogous effects of TTR aggregates on placental vasculature, including the impairment of capillary tube formation and induction of the anti-angiogenic factors sFlt-1 and sEng, have been described by Kalkunte and al [
11]. TTR mutations like V30M or age-driven misfolding and aggregation of wild-type TTR are involved in familial amyloid polyneuropathy (FAP) and senile systemic amyloidosis (SSR), respectively [
129]. Both disorders are characterized by the deposition of TTR fibrils in the peripheral nervous system in the case of FAP and, primarily, in cardiac tissue for SSR. The pathophysiology of these disorders mimics that of other proteinopathies, including the characteristic tissue and cellular damage inflicted by toxic oligomeric intermediates [
131].
5. Driving Mechanisms of Protein Misfolding in PE
5.1. ER Stress and Hyperactivation of the Unfolded Protein Response (UPR)
ER stress is a well-defined feature of PE [
23,
35,
60]. It is also a leading pathophysiologic process in protein conformational disorders [
137]. The ER is a focal site of protein synthesis and maturation. Following mRNA translation by ribosomes in the cytoplasm, peptides are translocated to the ER lumen, where they undergo necessary post-translational modifications, folding, and assembly into their functional conformation. To carry out these functions properly, the microenvironment in the ER lumen must maintain a high concentration of Ca
2+ and a high oxidative capacity [
60]. In PE, chronic low oxygen tension in the placenta, as a result of impaired uteroplacental perfusion, disrupts the delicate balance of the ER milieu, therefore inhibiting proper function [
60,
109]. This state of ER stress leads to the accumulation of unfolded proteins, which are more prone to misfold and aggregate, as the ER functional capacity is diminished [
138]. Common ER stress markers, such as ATF6, GRP78 (BiP/HSPA5), Ire1α, CHOP, PERK, and eIF2α, are upregulated in the PE placenta [
109,
134,
138,
139,
140,
141,
142,
143].
While UPR activation normally acts as a protective mechanism, an excessive and prolonged UPR, as would be the case during chronic tissue hypoxia, is self-damaging. Ultimately, cellular response mechanisms can be overwhelmed, leading to a collapse of protective systems and dooming cells for apoptosis.
5.2. Failure of the Protein Folding Machinery: The Role of Chaperones
Transcriptional upregulation of chaperones and folding enzymes is one of the compensatory mechanisms that are enacted by the UPR response. Under normal physiologic conditions, chaperones are molecules that aid other proteins in acquiring their natural conformation. They also have the capacity to recognize and bind to misfolded proteins, hence inhibiting or undoing their aggregation; and the capacity to unfold and refold improperly folded polypeptides, or tag them for degradation [
61]. ER stress is not the only inducer of chaperone activity. For example, heat shock proteins (HSPs) are a ubiquitously expressed family of chaperones that are upregulated in response to a variety of stressors that include heat, oxidative stress, sterile inflammation, and viral infection [
148,
149]. Members of the HSP70 family, in particular, are notoriously involved in the regulation of decidual and placental cell function from placentation to the end of pregnancy [
150]. Their upregulation in PE not only serves as a marker of UPR activation but also indicates that their overactivation could be driving pathological processes. If misfolded proteins are present in excess, increased expression of HSP70 isoforms like GRP78 is necessary for re-folding and disaggregating already tangled proteins. On the other hand, when bound to misfolded proteins, they are no longer available to perform other functions that are critical to maintaining cell integrity. This has been proposed as another possible mechanism driving the toxicity of protein aggregates like polyglutamine, an amyloidogenic protein involved in Huntington’s disease pathogenesis [
151].
In PE-associated misfolding, pregnancy zone protein (PZP) has also emerged as an important player. In contrast with GRP78, which is an ER-associated intracellular chaperone, PZP has recently been characterized as an extracellular chaperone that can inhibit the aggregation of Aβ [
135]. PZP is a dimeric homolog of the alpha-2-macroglobulin (α2M) that is found in high concentrations in the serum of pregnant women while levels are characteristically low in non-pregnant women and men [
152,
153,
154]. With a 71% degree of homology between the two protein sequences, it has been proposed that they may perform similar functions [
152,
155].
Plasminogen activator inhibitor type 2 (PAI-2), another extracellular chaperone, has been linked to PE-associated protein aggregation [
136]. Similarly to PZP, PAI-2, also known as SERPIN2B, is normally upregulated in pregnancy [
166]. PAI-2 plays an important role in inflammation through the regulation of T-helper-cell activation [
167]. The protein is expressed by activated macrophages but also endothelial cells and placental trophoblasts [
166]. With data indicating an association between reduced PAI-2 mRNA and protein levels, increased Th1 activation, placental insufficiency, and endothelial dysfunction in PE, PAI-2 has been proposed as a potential contributor to pathogenesis in pregnancy. [
52,
167,
168,
169]. The serpin can also act as a holdase, a type of chaperone protein that can passively bind to prefibrillar protein species, preventing them from further aggregation [
166,
170,
171].
5.3. Failure in Clearance Mechanisms: Defective Autophagolysosomal Processing
The third level at which disruptions in proteostasis mechanisms can occur is after all attempts at slowing the production of misfolded proteins and enhancing folding mechanisms have failed. At that time, misfolded proteins that exceed the UPR’s corrective capacity are targeted for degradation. Several studies and reviews have examined defects in autophagy as a potential root cause of protein aggregate accumulation in PE [
14,
122,
134,
181,
182,
183,
184]. Autophagy is one of two processes through which cells clear defective proteinssuch as aggregated proteins, and, damaged intracellular components like organelles [
63]. This process is mediated by a degradative enzyme in the lysosomes after the organelle fuses with autophagosomes carrying damaged cellular components (macroautophagy) or after the direct intake of individual unfolded proteins that were preemptively tagged (by ubiquitination) for degradation by chaperones (microautophagy) [
185]. The second mechanism through which intracellular proteolysis occurs is through the ubiquitin–proteasome pathway. Upon ubiquitination, proteins may diverge to the proteasome, a large protein complex that can hydrolyze and degrade proteins [
186].
Nakashima et al. postulated a decade ago that autophagy induction is critical to protecting placental trophoblast cells from hypoxia-induced disruptions. They showed that relative to autophagy-competent cells, autophagy-deficient trophoblast cells exhibited poor invasion and decreased ability to induce vascular remodeling in response to hypoxia, suggesting that defects in autophagy may contribute to poor vascular remodeling [
184,
187].
Cheng et al. showed that both the blockade of autophagosome–lysosome fusion with chloroquine and that of proteasome activity via MG132 treatment cause the accumulation of TTR aggregates in PHTs [
134,
188]. Moreover, there was an irregular placental accumulation of ubiquitinated proteins in the setting of EOPE-associated ER stress and UPR. These results, while preliminary, support that the impairment of autophagy, but also of ubiquitination-mediated protein degradation processes, may play a role in PE-associated protein aggregation [
134].
6. New Prediction, Diagnostic, and Treatment Approaches Based on PE-Associated Protein Misfolding
6.1. Early Prediction and Diagnosis
To date, accurate and timely PE diagnosis continues to pose a major challenge in clinical practice. The recent exclusion of proteinuria as a compulsory diagnostic criterion was favorable for the detection of non-proteinuric PE cases but left the presence of elevated blood pressure as the only sign that can be used to initiate further [
19,
197]. Based on current clinical diagnostic guidelines, the syndrome can only be detected when overt symptoms are already present.
Methods that have been developed in AD can inform us of the potential of misfolded protein detection to accurately predict and diagnose proteinopathies. For example, plasma and CSF assays assessing Aβ42/Aβ40 and tau/P-tau levels among other biomarkers are highly successful at identifying the presence of AD pathology prior to the threshold for amyloid positivity on PET scans and the onset of cognitive decline and at predicting disease trajectory [
199,
200,
201]. The biomarker assays perform even better when combined with genetic risk score [
202]. APOE-ε4, one of three alleles of the APOE gene, is well-established as the greatest genetic risk factor for AD. Adding APOE-ε4 status alone to prediction models increases AUC and, thus, predictive value [
202,
203].
A novel blood-based assay that can detect the presence of misfolded Aβ, TTR, or P-tau in the maternal circulation has been proposed by Cheng et al. [
15]. This method uses an autophagy-deficient trophoblast cell model to establish whether misfolded proteins are present in serum. The exposure of these specific autophagy-deficient human trophoblasts (ADTs) to serum from patients with PE, AD, and mild cognitive impairment leads to the intracellular accumulation of protein aggregates made of TTR and Aβ in PE cases, with the addition of α-synuclein in AD and MCI cases [
15].
The Congo red dot (CRD) test represents a non-invasive and low-cost alternative to other methods of assessing protein misfolding load associated with PE. The clinical applicability of the CRD test, which is based on the detection of urine congophilia, has been examined in fourteen studies, including one randomized control trial and one meta-analysis ([
90,
91,
204,
205,
206,
207,
208,
209,
210,
211,
212,
213,
214,
215]).
On the other hand, a few studies report low test performance. Results from Sammar et al. showed that the CRD test can diagnose PE with high accuracy in women presenting with symptoms; however, that the test is less effective at predicting PE in the first trimester of pregnancy when used alone [
208]. The predictive value of the test was improved when %CRR was used in combination with other markers of high disease risk, such as previous PE diagnosis, black race, body mass index, and mean arterial pressure [
208].
6.2. Treatment
Other than the emergent delivery of the fetus and placenta, which comes with its own set of iatrogenic complications when conducted preterm, there is no effective therapy for PE. Novel strategies that can be employed to reduce the accumulation of misfolded proteins and prevent their toxic effects include the inhibition of ER stress, stabilization of chaperone function, or restoration of autophagic balance. For example, the stabilization of TTR’s tetrameric structure is currently being explored as a therapeutic mechanism for the treatment of FAP and AD. Dissociation of the tetramer into monomers potentiates TTR’s aggregation into fibrils in FAP and compromises TTR’s ability to sequester unstable Aβ conformations in AD [
129,
132,
216,
217].
ER stress modulators, autophagy-enhancing drugs, gene therapy, and anti-amyloid immunotherapy are also being examined as therapeutic approaches in the treatment of AD [
146,
220,
221,
222]. In recent years, sildenafil, a known inducer of autophagy, has been associated with the alleviation of cognitive impairment in pre-clinical models and decreased incidence of AD in human studies and, thus, has been proposed as a candidate drug that could be repurposed for AD treatment [
223,
224]. The potential of sildenafil use for the treatment of AD has not yet been verified as evidence from a recent study does not support previous claims [
225].
7. Long-Term Implications of Pregnancy-Related Protein Misfolding Disease on Maternal and Fetal Health
As the field progresses in its understanding of PE-associated disease processes, the more complex the etiology of the disorder seems to become. Current evidence seems to point to a multisystem multifactorial pathological process, which extends beyond placental dysfunction. Gaining a better understanding of protein misfolding mechanisms and how they may contribute to systemic dysfunction in PE is crucial to advancing the ability to care for mothers and their babies during pregnancy. On the other hand, it also gives us a window into their future health. It is well-established that having PE during pregnancy greatly increases the risk of cardiovascular disease later in life for both mothers and their offspring [
237,
238,
239,
240]. Accumulating epidemiological reports also suggest a link between hypertensive disease during pregnancy and long-term risk of vascular dementia and AD [
241,
242].
While the discovery of significant protein misfolding pathology in PE opens many new avenues for investigation, diagnosis, and treatment, many questions are left to be answered. The central questions that persist and still require attention include whether protein aggregation instigates PE or is a consequence of it; the reasons behind the selective detection of certain proteins in specific tissues (serum vs. placenta vs. urine); and the systemic factors that may affect the accumulation and deposition of misfolded proteins in pregnancy. The possibility that proteostasis disruption might occur systemically rather than being confined at the placental level carries significant implications for both immediate and long-term maternal and fetal health.
This entry is adapted from the peer-reviewed paper 10.3390/molecules29030610