Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martin Tolar | -- | 3101 | 2024-03-20 15:53:46 | | | |
| 2 | Rita Xu | -234 word(s) | 2867 | 2024-03-21 03:06:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tolar, M.; Hey, J.A.; Power, A.; Abushakra, S. The Single Toxin Origin of Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/56439 (accessed on 10 August 2026).
Tolar M, Hey JA, Power A, Abushakra S. The Single Toxin Origin of Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/56439. Accessed August 10, 2026.
Tolar, Martin, John A. Hey, Aidan Power, Susan Abushakra. "The Single Toxin Origin of Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/56439 (accessed August 10, 2026).
Tolar, M., Hey, J.A., Power, A., & Abushakra, S. (2024, March 20). The Single Toxin Origin of Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/56439
Tolar, Martin, et al. "The Single Toxin Origin of Alzheimer’s Disease." Encyclopedia. Web. 20 March, 2024.
Copy Citation
New data suggest that the aggregation of misfolded native proteins initiates and drives the pathogenic cascade that leads to Alzheimer’s disease (AD) and other age-related neurodegenerative disorders. Researchers propose a unifying single toxin theory of brain neurodegeneration that identifies new targets and approaches to the development of disease-modifying treatments. An extensive body of genetic evidence suggests soluble aggregates of beta-amyloid (Aβ) as the primary neurotoxin in the pathogenesis of AD. New insights from fluid biomarkers, imaging, and clinical studies provide further evidence for the decisive impact of toxic Aβ species in the initiation and progression of AD.
Alzheimer’s disease
neurodegeneration
disease modification
beta-amyloid oligomers
APOE4
ALZ-801
valiltramiprosate
aducanumab
lecanemab
donanemab
1. Introduction
The past decade has brought remarkable advances in the diagnosis and understanding of the pathogenesis of Alzheimer’s disease (AD) and other neurodegenerative disorders, leading to the approval of the first wave of disease-modifying treatments. AD has become a model for the study of the origins and causes of brain neurodegeneration, from the use of fluid and imaging biomarkers tracking the progression of underlying pathologies to the application of such insights for successful drug development. Despite numerous studies evaluating a wide range of pharmacological treatments and drug mechanisms, only agents that block or prevent the formation of soluble beta-amyloid (Aβ, amyloid) aggregates called oligomers and protofibrils, or preferentially clear these species have shown clinical and biomarker efficacy in AD disease modification trials [1][2][3][4][5].
The Aβ peptide is a proteolytic derivative of the large transmembrane amyloid precursor protein (APP) and is formed by the sequential enzymatic cleavage of APP [6]. In its monomeric form, Aβ plays an important physiological role, protecting the human brain from injury, infection, and stress [7]. As a response to injury or stress, APP and Aβ production are upregulated, and Aβ concentrations, mostly Aβ40, increase acutely [8], providing the first-line response to infectious and toxic-metabolic brain insults. However, these responses, if left unchecked in the setting of deficient Aβ clearance mechanisms that occur with aging, can lead to Aβ accumulation and aggregation.
When brain concentrations of Aβ are elevated due to increased production as a result of genetic mutations, in response to injury or stressful conditions, or due to decreased brain clearance associated with aging, the monomeric Aβ peptides misfold and aggregate, gaining prion-like capabilities that seed further aggregation and propagation through brain structures [9]. Aβ peptides are inherently unstable and prone to misfolding and aggregation, especially the longer pathological Aβ42 species [10]. Aβ aggregation results in oligomers of many sizes, from dimers to dodecamers with molecular weights of 8–10 kDa to 60–80 kDa. Further Aβ aggregation yields larger oligomers with molecular weights of 80–500 kDa, called protofibrils, which are thought to be less toxic than the smaller oligomers. Misfolded Aβ propagates and spreads through the brain in a predictable and conserved pattern [10][11][12]. These oligomers and protofibrils inhibit long-term potentiation, the neuronal substrate for memory; cause neuronal stress and synaptic dystrophy; and trigger tau pathology that spreads along neuronal networks [11], ultimately leading to neuronal cell death [1][13]. The progressive synaptic failure and network dysfunction manifest as the progressive loss of cognitive abilities and executive function, described clinically as AD dementia.
One of the brain’s primary defenses against the toxicity of soluble oligomers is to sequester these neurotoxic species into insoluble amyloid fibrils and plaques, the pathognomonic histopathologic feature of AD [1][13]. Microglia, the immune cells of the brain, play a major role in the compaction of Aβ aggregates into insoluble and less toxic mature fibrils and dense plaques [14][15]. Additionally, perivascular astrocytes play a vital role in the clearance of Aβ aggregates into the glymphatic system and systemic circulation. At more advanced stages of the disease, microglia and astrocytes may also play a pro-inflammatory, harmful role in AD.
The primary risk factor for sporadic late-onset AD and many other neurodegenerative disorders is aging. In sporadic AD, the most common cause of the pathological increase in brain Aβ is impaired glymphatic clearance associated with aging due to either arteriosclerosis of the small brain vessels or the progressive dysfunction of the blood–brain barrier (BBB), affecting perivascular astrocytes and their aquaporin-4 water channels [16].
The second greatest risk factor for sporadic late-onset AD is the ε4 allele of apolipoprotein E (APOE4). APOE4 heterozygotes have an approximately four-fold increased risk of AD, whereas APOE4/4 homozygotes have a 10-fold to 12-fold increased risk compared with those with the neutral APOE3/3 genotype. The APOE4 genotype in AD is associated with accelerated Aβ aggregation, impaired Aβ clearance and an earlier age of AD onset due to insufficient uptake and clearance of Aβ through the LRP1 receptor on microglia and perivascular astrocytes [17], leading to deficiency in both intracellular proteolysis and glymphatic clearance of Aβ. APOE4 carriers accumulate more amyloid pathology and do so at a faster rate than non-carriers [18][19][20]. Recent proteomic analysis confirmed that APOE4 carriers have prominent dysfunction of the BBB [21].
Similarly, familial AD is caused by an increase in the production of Aβ due to mutations in APP or presenilin 1 or 2, leading to a very early onset of AD brain pathology and clinical symptoms in the fifth, fourth, or even third decade of life [22]. Another genetic form of AD is Down syndrome (DS) dementia, caused by the presence of the APP gene located on each chromosome 21, which is triplicated in DS. As a result, Aβ production is increased in DS, and characteristic AD brain pathology and biomarkers, as well as progressive cognitive impairment, are observed in most individuals with DS by the age of 40 [23][24].
AD has a very long preclinical phase, with a gradual accumulation of amyloid-driven pathology over approximately 20 years [25], followed by the phosphorylation of neuronal cytoskeletal microtubule protein tau. Elevated phosphorylated tau protein (p-tau) in the cerebrospinal fluid (CSF) or plasma is a marker of neuronal stress and a harbinger of incipient symptomatic AD.
Soluble tau “seeds” behave like prions and spread along synaptic networks, leading to their dysfunction. Soluble tau eventually aggregates into neurofibrillary tangles inside neurons, the second pathological hallmark of AD. This cascade of events, which is driven by increasing concentration of amyloid oligomers, is shown in Figure 1.
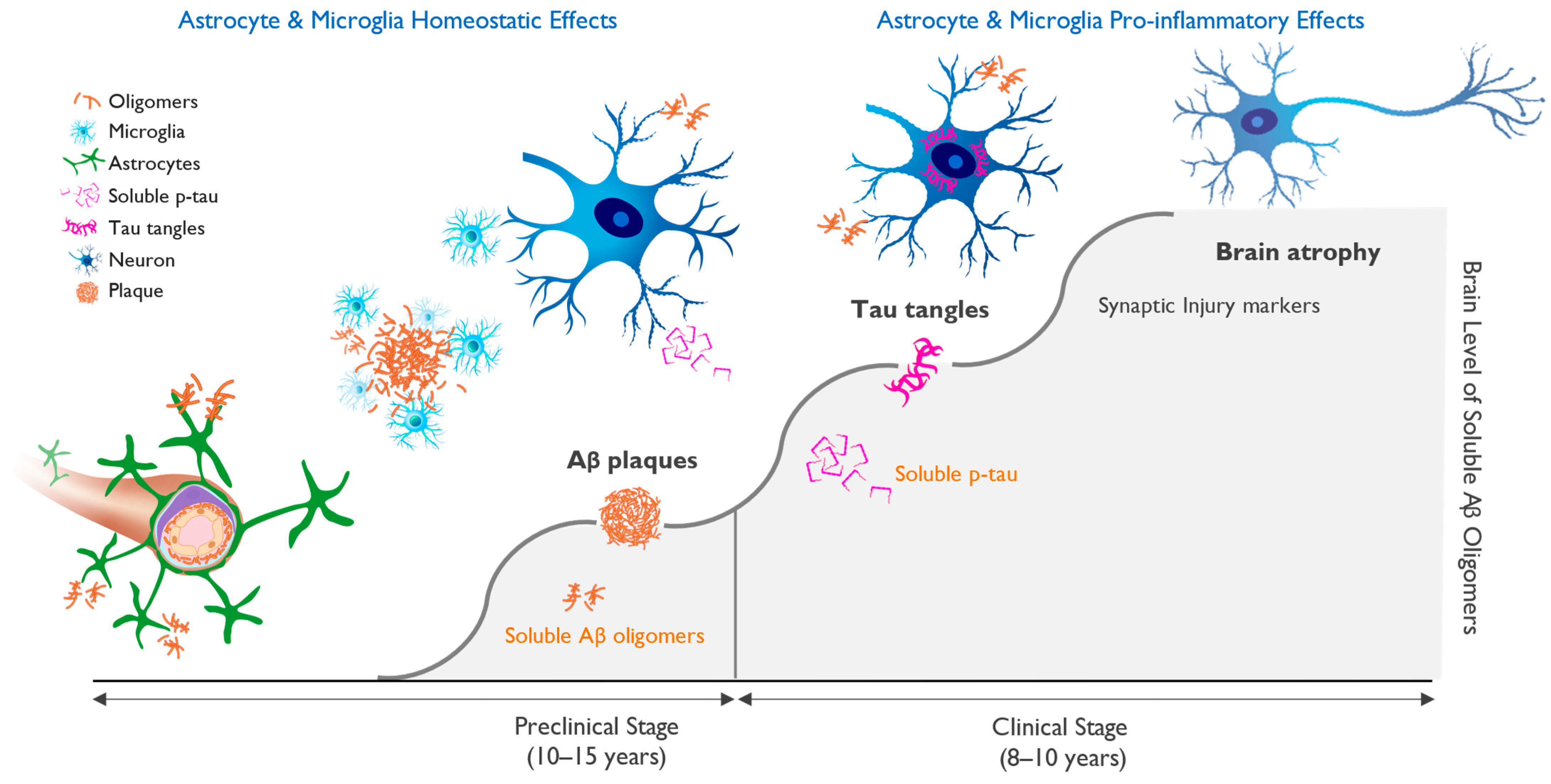
Figure 1. Progression of molecular pathology and neuronal dysfunction leading to clinical Alzheimer’s disease (AD). Clinical AD is preceded by a long silent pre-symptomatic phase. The accumulation of beta-amyloid-driven pathological changes in the brain occurs over 15–20 years and starts with the misfolding and aggregation of amyloid monomers into neurotoxic soluble oligomers, followed by neuronal dysfunction and cognitive impairment. Amyloid plaques serve as a protective brain mechanism, but once the ability to sequester beta-amyloid (Aβ) oligomers into insoluble fibrils and plaques is saturated, the oligomer toxicity triggers progressive neuronal stress with hyperphosphorylation of tau and the appearance of aggregated tau in neuronal cell bodies. This process correlates with markers of neuronal injury, eventually leading to neuronal cell loss and brain atrophy and the appearance of cognitive deficits. Figure 1 illustrates the importance of early diagnosis and intervention, ideally in the preclinical phase, in which treatment may allow for the maintenance of brain health and normal brain function.
Amyloid and tau pathologies are the two defining features of AD, and the current biological basis of AD diagnosis for clinical trials requires positive amyloid and tau confirmation [12]. The earliest clinical stage of AD is defined as mild cognitive impairment (MCI), which progresses into mild AD once functional deficits appear several years later.
2. APOE4 Represents Main Genetic Risk Factor for Alzheimer’s Disease
Compared to APOE4 noncarriers, the risk of AD is four-fold higher for APOE4 heterozygotes and 10-fold to 12-fold higher for APOE4/4 homozygotes [26] in populations based only on clinical diagnosis, rising to odds ratios of 4.6 and 25.4, respectively, for AD subjects diagnosed using CSF biomarkers [27]. As a result, approximately two-thirds of AD patients are carriers of the APOE4 gene, with roughly 50% being APOE4 heterozygotes and 15% being APOE4/4 homozygotes, as shown in Figure 2.
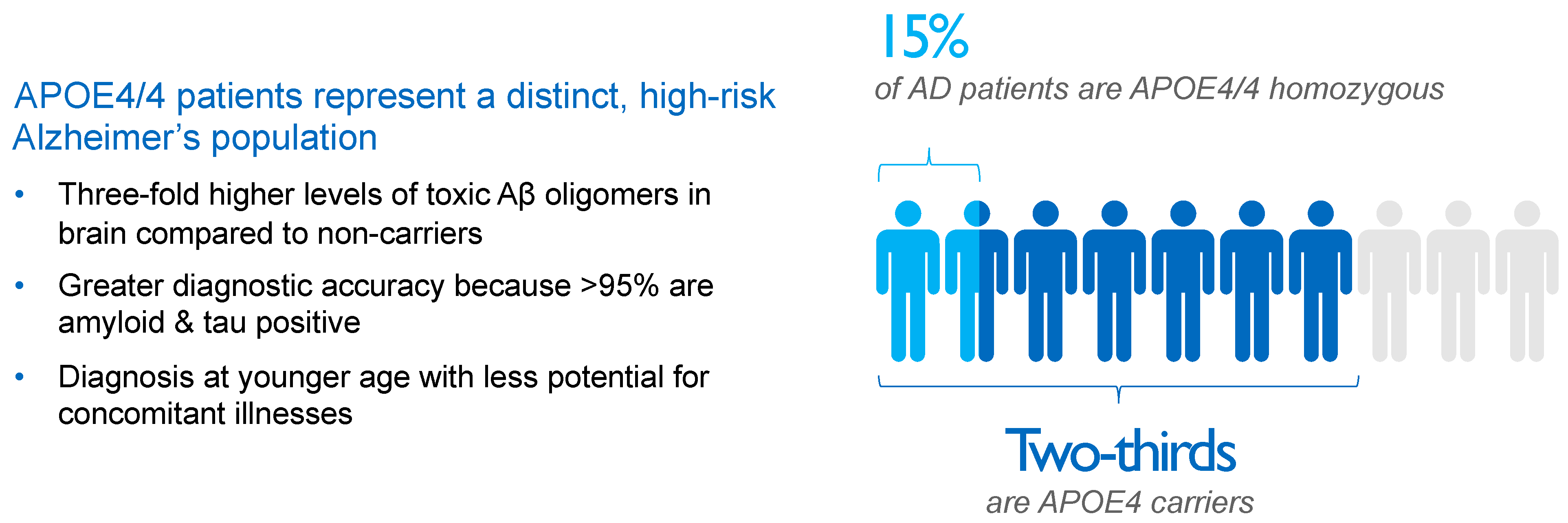
Figure 2. Impact and distribution of APOE4 genotypes in Alzheimer’s disease (AD) patients. APOE4 carriers represent approximately two-thirds of AD patients. APOE4/4 homozygotes, patients with the most aggressive form of sporadic AD, show several-fold higher concentrations of amyloid oligomers in the brain compared with non-carriers and an almost decade-earlier onset of the disease, thereby representing a suitable high-risk population to study the effects of anti-amyloid interventions. Aβ, beta-amyloid; AD, Alzheimer’s disease; APOE4, ε4 allele of apolipoprotein E gene; APOE4/4, homozygosity for ε4 allele of apolipoprotein E gene.
APOE is a lipoprotein that transports cholesterol and Aβ in the brain and plasma. Approximately 25% of individuals in the Caucasian population carry one APOE4 allele, and 2% to 3% of the population carries two alleles, but these APOE4 carriers comprise 65% to 70% of AD subjects in clinical trials utilizing a biomarker-based diagnosis of AD [27]. Approximately 10% to 15% of the 6.7 million AD patients in the United States are APOE4/4 homozygotes, with a similar number of APOE4/4 homozygotes with AD (~700,000) in the European Union [28][29].
As a result of increased brain Aβ concentration, APOE4 carriers develop more vascular amyloid pathology [30][31], called cerebral amyloid angiopathy (CAA). The deposition of amyloid in brain microvasculature and the resultant weakening of vessel walls underlies the occurrence of spontaneous brain edema and microhemorrhages observed on magnetic resonance imaging (MRI) scans [32], as well as the risk of brain edema and microhemorrhages following treatment with anti-amyloid antibodies [2]. These MRI lesions, when they occur via treatment with amyloid immunotherapies, are known as amyloid-related imaging abnormalities either with edema (ARIA-E) or with microhemorrhages and/or hemosiderin deposits (ARIA-H).
ARIA lesions were first described in the bapineuzumab trials [33], and, subsequently, they were reported in trials with other plaque-clearing antibodies [2][34][35]. APOE4/4 homozygous AD patients show the highest degree of CAA pathology [36] and also carry the highest risk of ARIA-E and ARIA-H when treated with anti-amyloid antibodies [2][35]. Therefore, there is an urgent unmet medical need for an agent that can deliver meaningful clinical efficacy in APOE4/4 homozygotes, without the increased risk of brain edema or microhemorrhage.
Since the oral anti-amyloid oligomer agent ALZ-801/valiltramiprosate inhibits the formation of Aβ oligomers without affecting plaques [37], ALZ-801 has the potential to be a suitable therapeutic for APOE4 carriers that does not increase the risk of ARIA.
3. Interaction of Aβ and APOE4 with Other Molecules in AD Brain
The interaction of Aβ species with various proteins in the AD brain is an area of increasing importance, and studies of the Aβ interactome have the potential to provide novel therapeutic targets [38]. One of the most studied protein interactions in AD is that of Aβ and APOE4 [39][40]. There are multiple mechanisms by which the APOE4 genotype confers increased AD risk, including decreased Aβ phagocytosis and clearance through the blood–brain barrier, increased tau hyperphosphorylation and aggregation, and exaggerated microglial and astrocytic responses. These effects collectively lead to diminished Aβ clearance, exaggerated neuroinflammation, and increased Aβ aggregation [41][42], all leading to increased levels of neurotoxic soluble Aβ oligomers and tau spreading in the brains of APOE4 carriers, especially APOE4/4 homozygotes [43]. Consistent with having the greatest amyloid burden and tau pathology, APOE4/4 subjects exhibit an earlier and faster rate of cognitive decline, becoming symptomatic approximately a decade earlier than non-carriers.
The interaction of APOE4 with the microglial activating receptor TREM2 is an emerging area of focus for drug discovery [44][45]. This is a complex relationship, with the effects of microglial activation being homeostatic and protective in the early stages of AD and pro-inflammatory and harmful at later stages. Therefore, TREM2-targeted drug development is challenging since the benefits of TREM2 activation may be specific to the disease stage.
Recent studies have highlighted the APOE4 interaction with the Reelin-Disabled-1 (Dab1) signaling pathway, which plays a protective role in synaptic development and plasticity [46]. Reelin signaling protects synapses from Aβ-induced neuronal stress, while APOE4 interferes with this protective effect, worsening synaptic toxicity of Aβ [47]. Genetic variations in the Reelin pathway have been described in a Spanish population to confer increased risk [48] and in the UK Biobank as risk factors for AD, particularly in APOE4/4 homozygotes [49]. A recent report described a protective Reelin variant in a male with autosomal-dominant AD [50].
4. Biomarkers and Biological Definition of Alzheimer’s Disease
The biological definition of AD has been essential for the success of drug trials with disease-modifying agents that target amyloid pathology, the only approach to date that has shown positive clinical and biomarker effects in delaying the progression of the disease. A clinical AD diagnosis without biomarker confirmation has very low accuracy in APOE4 non-carriers (~60%) and low accuracy in heterozygotes (80%) but has excellent accuracy in APOE4/4 homozygotes (>95%) [51].
New insights into the pathogenic role of Aβ and the application of brain biomarkers have markedly improved our ability to identify individuals with AD pathology long before the onset of clinical symptoms and have led to diagnostic criteria for clinical research based on objective disease biomarkers [12]. Data from longitudinal AD studies and interventional clinical trials using PET imaging of amyloid and tau aggregates and fluid biomarkers of AD were most helpful in advancing a biological definition of AD that avoids the pitfalls and substantial inaccuracy of reaching a clinical diagnosis, which was 30% in older clinical trials [12][51]. The onset of biomarker changes closely correlates with clinical onset and stages of AD and follows a well-defined sequence of pathological changes. An increase in brain Aβ concentration leads to amyloid accumulation into toxic soluble oligomers and protofibrils, initiating tau phosphorylation that can be detected by amyloid and tau PET scans. An increase in CSF or plasma biomarkers, including Aβ, p-tau, and neurofilament light chain protein (NfL), precedes brain volume loss that can be assessed by brain volumetric MRI and ultimately results in cognitive decline [52][53][54][55].
Biomarker-based AD diagnostic criteria hold the potential to enable the detection of pathological changes years and decades before the onset of clinical symptoms and require that subjects have positive amyloid and tau biomarkers, with or without evidence of neuronal injury or neurodegeneration. A/T/N classification is based on the status of amyloid (A), tau (T), and neuronal pathology (N), as determined by amyloid and tau PET imaging or CSF or plasma biomarkers, including Aβ, p-tau-, NfL, and neurogranin, and by MRI measurements of brain volume loss focused on hippocampal volume and cortical thickness.
P-tau has recently emerged as the most reliable diagnostic and staging marker in AD. Tau, a cytoskeletal protein that forms the scaffolding of neurons called microtubules, is phosphorylated at threonine 181 or 217 sites when neurons are stressed and injured by toxic amyloid oligomers [52][53][56]. Longitudinal studies in AD subjects have shown that as levels of aggregated forms of amyloid increase in the brain, they induce abnormal phosphorylation of neuronal tau and a progressive elevation of p-tau in the CSF and plasma. These progressive increases in CSF and plasma p-tau precede the appearance of intraneuronal neurofibrillary tangles, the tau pathology that can be detected by tau PET imaging [54]. An increase in the concentration of toxic amyloid oligomers induces downstream synaptic dysfunction and neuronal injury, leading to the formation of p-tau, as shown in Figure 3.
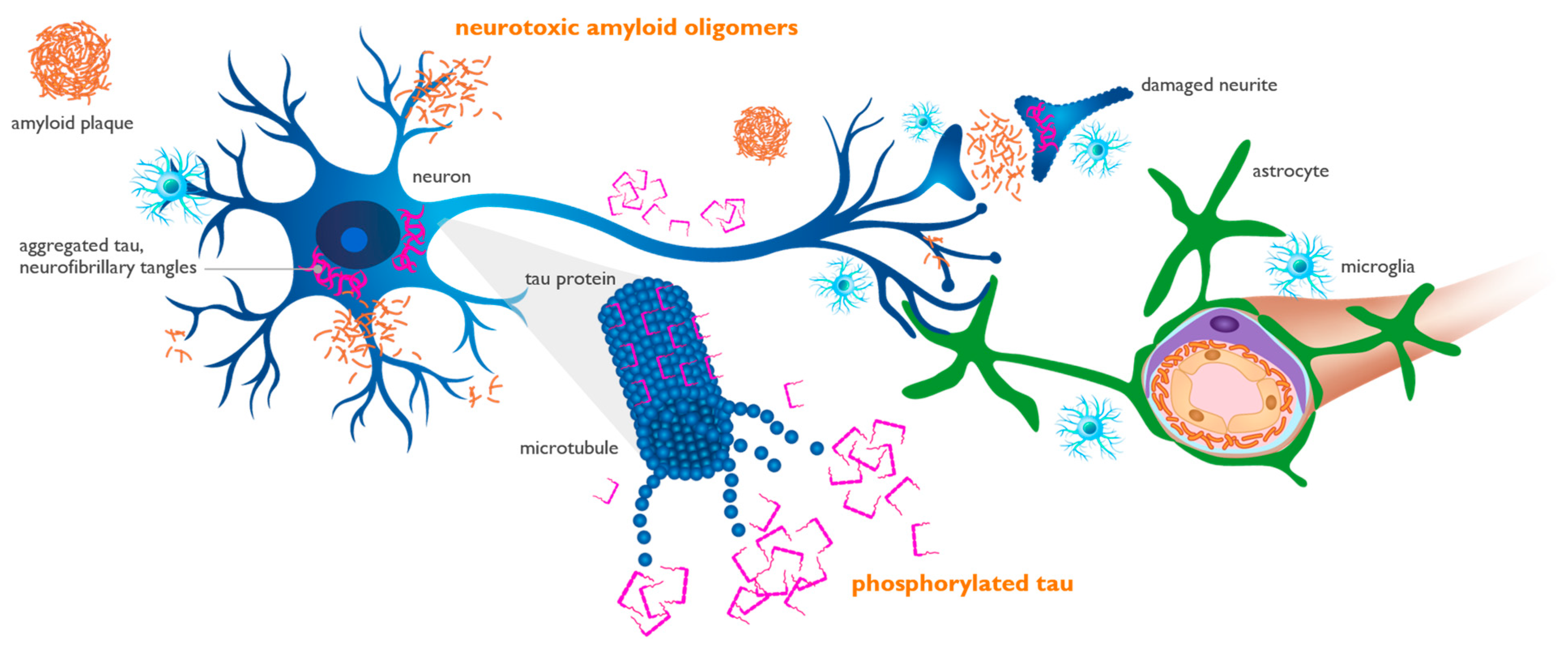
Figure 3. Amyloid oligomers induce synaptic dysfunction and neuronal injury, leading to the phosphorylation of tau protein. Amyloid oligomers injure synapses located on neuronal dendrites and cell bodies (blue cell). Phosphorylated tau (p-tau) is released from injured neurons into the interstitial fluid and can be measured in the cerebrospinal fluid and plasma, representing neuronal dysfunction and loss. Aggregated tau (purple) is shown as twisted neurofibrillary tangles within neurons, forming tau tangles. Perivascular astrocytes (green cell) play an active role in the trafficking and clearance of beta-amyloid (Aβ) and p-tau through the glymphatic perivascular system into systemic circulation, where they can be detected by plasma assays.
P-tau181 and p-tau217 isoforms are found in the brain, CSF, and plasma of patients with AD pathology decades before clinical onset and have been shown to appear as a response to neurotoxic soluble amyloid aggregates [54][55]. These p-tau isoforms are specific to AD and serve as reliable biomarkers for tracking disease progression and for determining the impact of effective anti-amyloid therapeutics [52][53][55].
CSF assays for core AD biomarkers have been continually optimized, and some are approved for clinical use. More recently, plasma biomarker assays have also become sensitive and reliable for use in clinical trials. The late-stage amyloid antibody trials have included the evaluation of plasma biomarkers [3][4][57][58], focusing on p-tau181 and p-tau217 isoforms, which have been associated with efficacy on standard clinical endpoints.
Anti-amyloid antibodies with meaningful clinical efficacy show plasma p-tau181 reductions of ≥15% from baseline over 1 year, while those that failed to achieve efficacy reduced p-tau181 by <10% over 1 year. This suggests that (1) the magnitude of plasma p-tau181 reduction over 1 year is a reasonable predictor of clinical efficacy and (2) early and sustained reduction in p-tau181 may be a suitable marker of target engagement and meaningful clinical efficacy. Lecanemab also showed a reduction in the synaptic injury marker neurogranin in the CSF, whereas NfL levels in the CSF did not separate from the placebo over 78 weeks of treatment. Both lecanemab and donanemab showed a significant reduction in the astrocytic marker plasma glial fibrillary acidic protein (GFAP) over 78 weeks [4][57][59].
The dose of oral ALZ-801/valiltramiprosate being used in current clinical trials provides CNS concentrations that fully block the formation of neurotoxic soluble oligomers in mechanism of action studies [60]. This dose has shown the most pronounced p-tau181 reduction compared with the effects reported with other anti-amyloid agents, including anti-amyloid antibodies [61][62]. This is consistent with the promising clinical efficacy that has been observed in APOE4 carriers treated with ALZ-801′s active agent tramiprosate.
References
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560.
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95.
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210.
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Eng. J. Med. 2023, 388, 9–21.
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in early symptomatic Alzheimer disease The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 2023, 330, 512–527.
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235.
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell. Longev. 2014, 2014, 795375.
- Hefter, D.; Draguhn, A. APP as a protective factor in acute neuronal insults. Front. Mol. Neurosci. 2017, 10, 22.
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133.
- Duran-Aniotz, C.; Moreno-Gonzalez, I.; Gamez, N.; Perez-Urrutia, N.; Vegas-Gomez, L.; Soto, C.; Morales, R. Amyloid pathology arrangements in Alzheimer’s disease brains modulate in vivo seeding capability. Acta Neuropathol. Commun. 2021, 9, 56.
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and a-synuclein in neurodegeneration. Brain 2017, 140, 266–278.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive Alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355.
- Wang, Z.; Weaver, D.F. Microglia and microglial-based receptors in the pathogenesis and treatment of Alzheimer’s disease. Int. Immunopharmacol. 2022, 110, 109070.
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 2020, 40, 1956–1974.
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wan, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimers Res. Ther. 2022, 14, 59.
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303.
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938.
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid PET Study Group; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949.
- Tijms, B.M.; Vromen, E.M.; Mjaavatten, O.; Holstege, H.; Reus, L.M.; van der Lee, S.; Wesenhagen, K.E.J.; Lorenzini, L.; Vermunt, L.; Venkatraghavan, V.; et al. Cerebrospinal fluid proteomics in patients with Alzheimer’s disease reveals five molecular subtypes with distinct genetic risk profiles. Nat. Aging 2024, 4, 33–47.
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Eng. J. Med. 2012, 367, 795–804.
- Rafii, M.S.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; Lott, I.; Klunk, W.; Head, E.; Christian, B.; et al. The AT(N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement. 2020, 12, e12062.
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997.
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: Correlation with amyloid PET. Brain 2017, 140, 1486–1498.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923.
- Saddiki, H.; Fayosse, A.; Cognat, E.; Sabia, S.; Engelborghs, S.; Wallon, D.; Alexopoulos, P.; Blennow, K.; Zetterberg, H.; Parnetti, L.; et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020, 17, e1003289.
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 13–14.
- Ward, A.; Crean, S.; Mercaldi, C.J.; Collins, J.M.; Boyd, D.; Cook, M.N.; Arrighi, H.M. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: A systematic review and meta-analysis. Neuroepidemiology 2012, 38, 1–17.
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653.
- Chalmers, K.; Wilcock, G.K.; Love, S. APOE ε4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol. Appl. Neurobiol. 2003, 29, 231–238.
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42.
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011, 7, 367–385.
- Withington, C.G.; Turner, R.S. Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer’s disease. Front. Neurol. 2022, 13, 862369.
- Leqembi (lecanemab-irmb) Injection for Intravenous Use ; Eisai Inc.: Nutley, NJ, USA, 2023; Available online: https://www.leqembi.com/ (accessed on 14 September 2023).
- Abushakra, S.; Mandelbaum, R.; Barakos, J.; Scheltens, P.; Porsteinsson, A.P.; Watson, D.; MacSweeney, E.; Sabbagh, M.; Liang, E.; Kesslak, P.; et al. Prevalence of amyloid-related imaging abnormalities in APOE4/4 homozygotes with early Alzheimer’s disease (AD): Baseline findings from ongoing clinical trials of the oral anti-amyloid agent ALZ-801 (valiltramiprosate) (P5-6.003). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023.
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: Integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs 2017, 31, 495–509.
- Lazarev, V.F.; Dutysheva, E.A.; Kanunikov, I.E.; Guzhova, I.V.; Margulis, B.A. Protein interactome of amyloid-β as a therapeutic target. Pharmaceuticals 2023, 16, 312.
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. J. Mol. Biol. 2019, 431, 2248–2265.
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754.
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192.
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7.
- Gratuze, M.; Schlachetzki, J.C.M.; D’Oliveira Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O’Brien, S.; et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 2023, 111, 202–219.e7.
- Jolly-Amado, A.; Kulkarni, N.; Nash, K.R. Reelin signaling in neurodevelopmental disorders and neurodegenerative diseases. Brain Sci. 2023, 13, 1479.
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid β toxicity. Sci. Signal. 2015, 8, ra67.
- Bufill, E.; Roura-Poch, P.; Sala-Matavera, I.; Antón, S.; Lleó, A.; Sánchez-Saudinós, B.; Tomàs-Abadal, L.; Puig, T.; Abós, J.; Bernades, S.; et al. Reelin signaling pathway genotypes and Alzheimer disease in a Spanish population. Alzheimer Dis. Assoc. Disord. 2015, 29, 169–172.
- Bracher-Smith, M.; Leonenko, G.; Baker, E.; Crawford, K.; Graham, A.C.; Salih, D.A.; Howell, B.W.; Hardy, J.; Escott-Price, V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2022, 119, 67–76.
- Lopera, F.; Marino, C.; Chandrahas, A.S.; O’Hare, M.; Villalba-Moreno, N.D.; Aguillon, D.; Baena, A.; Sanchez, J.S.; Vila-Castelar, C.; Ramirez Gomez, L.; et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 2023, 29, 1243–1252.
- Degenhardt, E.K.; Witte, M.M.; Case, M.G.; Yu, P.; Henley, D.B.; Hochstetler, H.M.; D’Souza, D.N.; Trzepacz, P.T. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 2016, 57, 208–216.
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433.
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018, 14, 1470–1481.
- Mattsson-Calgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz2387.
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 2022, 28, 1797–1801.
- Blennow, K.; Zetterberg, H. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog. Mol. Biol. Transl. Sci. 2019, 168, 3–23.
- Pontecorvo, M.J.; Lu, M.; Burnham, S.C.; Schade, A.E.; Dage, J.L.; Shcherbinin, S.; Collins, E.C.; Sims, J.R.; Mintun, M.A. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: A secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022, 79, 1250–1259.
- Bittner, T.; Blennow, K.; Scelsi, M.; Palermo, G.; Kollmorgen, G.; Smith, J.; Zetterberg, H.; Doody, R.S. GRADUATE I and II Results. Effect of Subcutaneous Gantenerumab on Fluid Biomarkers of AD Pathology and Neurodegeneration with Insights on Plasma Biomarkers and New CSF Biomarkers. Available online: https://medically.gene.com/global/en/unrestricted/neuroscience/adpd-2023/adpd-2023-presentation-bittner-graduate-i-and-ii-result.html (accessed on 14 September 2023).
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; de Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta (1–42/1–40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res. Ther. 2020, 12, 118.
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs 2018, 32, 849–861.
- Abushakra, S.; Hey, J.; Blennow, K.; Scheltens, P.; Reiman, E.M.; Hort, J.; Sheardova, K.; Rutgers, S.M.; Prins, N.D.; Dautzenberg, P.; et al. Effects of oral ALZ-801, an amyloid oligomer inhibitor, on plasma biomarkers in APOE4 carriers with Early Alzheimer’s disease: Results of six-month interim analysis from a phase 2 biomarker study. Alzheimers Dement. 2022, 18, e069141. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/epdf/10.1002/alz.069141 (accessed on 14 September 2023).
- Hey, J.H.; Abushakra, S.; Blennow, K.; Scheltens, P.; Hort, J.; Sheardova, K.; Prins, N.D.; Rutgers, M.S.; Dautzenberg, P.L.; Pazdert, L.; et al. Effects of ALZ-801, an oral amyloid oligomer inhibitor, on biomarkers of Alzheimer’s disease (AD): 12-month results of phase 2 biomarker study in early AD (S26.0007). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023; Available online: https://www.aan.com/MSA/Public/Events/AbstractDetails/52624 (accessed on 14 September 2023).
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
21 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No