New data suggest that the aggregation of misfolded native proteins initiates and drives the pathogenic cascade that leads to Alzheimer’s disease (AD) and other age-related neurodegenerative disorders. ResWearchers propose a unifying single toxin theory of brain neurodegeneration that identifies new targets and approaches to the development of disease-modifying treatments. An extensive body of genetic evidence suggests soluble aggregates of beta-amyloid (Aβ) as the primary neurotoxin in the pathogenesis of AD. New insights from fluid biomarkers, imaging, and clinical studies provide further evidence for the decisive impact of toxic Aβ species in the initiation and progression of AD.
Understanding the distinct roles of soluble and insoluble amyloid aggregates on AD pathogenesis has been the key missing piece of the Alzheimer’s puzzle. Data from clinical trials with anti-amyloid agents and recent advances in the diagnosis of AD demonstrate that the driving insult in biologically defined AD is the neurotoxicity of soluble Aβ aggregates, called oligomers and protofibrils, rather than the relatively inert insoluble mature fibrils and amyloid plaques. Amyloid oligomers appear to be the primary factor causing the synaptic impairment, neuronal stress, spreading of tau pathology, and eventual cell death that lead to the clinical syndrome of AD dementia. All other biochemical effects and neurodegenerative changes in the brain that are observed in AD are a response to or a downstream effect of this initial toxic insult by oligomers.
Other neurodegenerative disorders follow a similar pattern of pathogenesis, in which normal brain proteins with important biological functions become trapped in the aging brain due to impaired clearance and then misfold and aggregate into neurotoxic species that exhibit prion-like behavior. These aggregates then spread through the brain and cause disease-specific neurodegeneration. Targeting the inhibition of this initial step in neurodegeneration by blocking the misfolding and aggregation of healthy proteins has the potential to slow or arrest disease progression, and if treatment is administered early in the course of AD and other neurodegenerative disorders, it may delay or prevent the onset of clinical symptoms.
1. Introduction
The past decade has brought remarkable advances in the diagnosis and understanding of the pathogenesis of Alzheimer’s disease (AD) and other neurodegenerative disorders, leading to the approval of the first wave of disease-modifying treatments. AD has become a model for the study of the origins and causes of brain neurodegeneration, from the use of fluid and imaging biomarkers tracking the progression of underlying pathologies to the application of such insights for successful drug development. Despite numerous studies evaluating a wide range of pharmacological treatments and drug mechanisms, only agents that block or prevent the formation of soluble beta-amyloid (Aβ, amyloid) aggregates called oligomers and protofibrils, or preferentially clear these species have shown clinical and biomarker efficacy in AD disease modification trials
[1][2][3][4][5][1,2,3,4,5].
The Aβ peptide is a proteolytic derivative of the large transmembrane amyloid precursor protein (APP) and is formed by the sequential enzymatic cleavage of APP
[6]. In its monomeric form, Aβ plays an important physiological role, protecting the human brain from injury, infection, and stress
[7]. As a response to injury or stress, APP and Aβ production are upregulated, and Aβ concentrations, mostly Aβ40, increase acutely
[8], providing the first-line response to infectious and toxic-metabolic brain insults. However, these responses, if left unchecked in the setting of deficient Aβ clearance mechanisms that occur with aging, can lead to Aβ accumulation and aggregation.
When brain concentrations of Aβ are elevated due to increased production as a result of genetic mutations, in response to injury or stressful conditions, or due to decreased brain clearance associated with aging, the monomeric Aβ peptides misfold and aggregate, gaining prion-like capabilities that seed further aggregation and propagation through brain structures
[9]. Aβ peptides are inherently unstable and prone to misfolding and aggregation, especially the longer pathological Aβ42 species
[10]. Aβ aggregation results in oligomers of many sizes, from dimers to dodecamers with molecular weights of 8–10 kDa to 60–80 kDa. Further Aβ aggregation yields larger oligomers with molecular weights of 80–500 kDa, called protofibrils, which are thought to be less toxic than the smaller oligomers. Misfolded Aβ propagates and spreads through the brain in a predictable and conserved pattern
[10][11][12][10,11,12]. These oligomers and protofibrils inhibit long-term potentiation, the neuronal substrate for memory; cause neuronal stress and synaptic dystrophy; and trigger tau pathology that spreads along neuronal networks
[11], ultimately leading to neuronal cell death
[1][13][1,13]. The progressive synaptic failure and network dysfunction manifest as the progressive loss of cognitive abilities and executive function, described clinically as AD dementia.
One of the brain’s primary defenses against the toxicity of soluble oligomers is to sequester these neurotoxic species into insoluble amyloid fibrils and plaques, the pathognomonic histopathologic feature of AD
[1][13][1,13]. Microglia, the immune cells of the brain, play a major role in the compaction of Aβ aggregates into insoluble and less toxic mature fibrils and dense plaques
[14][15][14,15]. Additionally, perivascular astrocytes play a vital role in the clearance of Aβ aggregates into the glymphatic system and systemic circulation. At more advanced stages of the disease, microglia and astrocytes may also play a pro-inflammatory, harmful role in AD.
The primary risk factor for sporadic late-onset AD and many other neurodegenerative disorders is aging. In sporadic AD, the most common cause of the pathological increase in brain Aβ is impaired glymphatic clearance associated with aging due to either arteriosclerosis of the small brain vessels or the progressive dysfunction of the blood–brain barrier (BBB), affecting perivascular astrocytes and their aquaporin-4 water channels
[16].
The second greatest risk factor for sporadic late-onset AD is the ε4 allele of apolipoprotein E (APOE4). APOE4 heterozygotes have an approximately four-fold increased risk of AD, whereas APOE4/4 homozygotes have a 10-fold to 12-fold increased risk compared with those with the neutral APOE3/3 genotype. The APOE4 genotype in AD is associated with accelerated Aβ aggregation, impaired Aβ clearance and an earlier age of AD onset due to insufficient uptake and clearance of Aβ through the LRP1 receptor on microglia and perivascular astrocytes
[17], leading to deficiency in both intracellular proteolysis and glymphatic clearance of Aβ. APOE4 carriers accumulate more amyloid pathology and do so at a faster rate than non-carriers
[18][19][20][18,19,20]. Recent proteomic analysis confirmed that APOE4 carriers have prominent dysfunction of the BBB
[21].
Similarly, familial AD is caused by an increase in the production of Aβ due to mutations in APP or presenilin 1 or 2, leading to a very early onset of AD brain pathology and clinical symptoms in the fifth, fourth, or even third decade of life
[22]. Another genetic form of AD is Down syndrome (DS) dementia, caused by the presence of the APP gene located on each chromosome 21, which is triplicated in DS. As a result, Aβ production is increased in DS, and characteristic AD brain pathology and biomarkers, as well as progressive cognitive impairment, are observed in most individuals with DS by the age of 40
[23][24][23,24].
AD has a very long preclinical phase, with a gradual accumulation of amyloid-driven pathology over approximately 20 years
[25], followed by the phosphorylation of neuronal cytoskeletal microtubule protein tau. Elevated phosphorylated tau protein (p-tau) in the cerebrospinal fluid (CSF) or plasma is a marker of neuronal stress and a harbinger of incipient symptomatic AD.
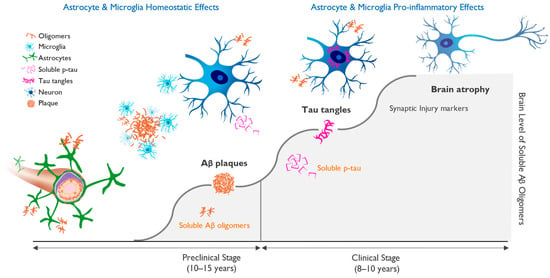
Soluble tau “seeds” behave like prions and spread along synaptic networks, leading to their dysfunction. Soluble tau eventually aggregates into neurofibrillary tangles inside neurons, the second pathological hallmark of AD. This cascade of events, which is driven by increasing concentration of amyloid oligomers, is shown in Figure 1.
Figure 1. Progression of molecular pathology and neuronal dysfunction leading to clinical Alzheimer’s disease (AD). Clinical AD is preceded by a long silent pre-symptomatic phase. The accumulation of beta-amyloid-driven pathological changes in the brain occurs over 15–20 years and starts with the misfolding and aggregation of amyloid monomers into neurotoxic soluble oligomers, followed by neuronal dysfunction and cognitive impairment. Amyloid plaques serve as a protective brain mechanism, but once the ability to sequester beta-amyloid (Aβ) oligomers into insoluble fibrils and plaques is saturated, the oligomer toxicity triggers progressive neuronal stress with hyperphosphorylation of tau and the appearance of aggregated tau in neuronal cell bodies. This process correlates with markers of neuronal injury, eventually leading to neuronal cell loss and brain atrophy and the appearance of cognitive deficits. Figure 1 illustrates the importance of early diagnosis and intervention, ideally in the preclinical phase, in which treatment may allow for the maintenance of brain health and normal brain function.
Amyloid and tau pathologies are the two defining features of AD, and the current biological basis of AD diagnosis for clinical trials requires positive amyloid and tau confirmation
[12]. The earliest clinical stage of AD is defined as mild cognitive impairment (MCI), which progresses into mild AD once functional deficits appear several years later.
2. APOE4 Represents Main Genetic Risk Factor for Alzheimer’s Disease
Compared to APOE4 noncarriers, the risk of AD is four-fold higher for APOE4 heterozygotes and 10-fold to 12-fold higher for APOE4/4 homozygotes
[26][64] in populations based only on clinical diagnosis, rising to odds ratios of 4.6 and 25.4, respectively, for AD subjects diagnosed using CSF biomarkers
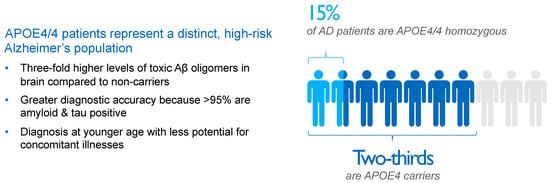
[27][65]. As a result, approximately two-thirds of AD patients are carriers of the APOE4 gene, with roughly 50% being APOE4 heterozygotes and 15% being APOE4/4 homozygotes, as shown in
Figure 2.
Figure 2. Impact and distribution of APOE4 genotypes in Alzheimer’s disease (AD) patients. APOE4 carriers represent approximately two-thirds of AD patients. APOE4/4 homozygotes, patients with the most aggressive form of sporadic AD, show several-fold higher concentrations of amyloid oligomers in the brain compared with non-carriers and an almost decade-earlier onset of the disease, thereby representing a suitable high-risk population to study the effects of anti-amyloid interventions. Aβ, beta-amyloid; AD, Alzheimer’s disease; APOE4, ε4 allele of apolipoprotein E gene; APOE4/4, homozygosity for ε4 allele of apolipoprotein E gene.
APOE is a lipoprotein that transports cholesterol and Aβ in the brain and plasma. Approximately 25% of individuals in the Caucasian population carry one APOE4 allele, and 2% to 3% of the population carries two alleles, but these APOE4 carriers comprise 65% to 70% of AD subjects in clinical trials utilizing a biomarker-based diagnosis of AD
[27][65]. Approximately 10% to 15% of the 6.7 million AD patients in the United States are APOE4/4 homozygotes, with a similar number of APOE4/4 homozygotes with AD (~700,000) in the European Union
[28][29][66,67].
As a result of increased brain Aβ concentration, APOE4 carriers develop more vascular amyloid pathology
[30][31][68,69], called cerebral amyloid angiopathy (CAA). The deposition of amyloid in brain microvasculature and the resultant weakening of vessel walls underlies the occurrence of spontaneous brain edema and microhemorrhages observed on magnetic resonance imaging (MRI) scans
[32][70], as well as the risk of brain edema and microhemorrhages following treatment with anti-amyloid antibodies
[2]. These MRI lesions, when they occur via treatment with amyloid immunotherapies, are known as amyloid-related imaging abnormalities either with edema (ARIA-E) or with microhemorrhages and/or hemosiderin deposits (ARIA-H).
ARIA lesions were first described in the bapineuzumab trials
[33][71], and, subsequently, they were reported in trials with other plaque-clearing antibodies
[2][34][35][2,72,73]. APOE4/4 homozygous AD patients show the highest degree of CAA pathology
[36][74] and also carry the highest risk of ARIA-E and ARIA-H when treated with anti-amyloid antibodies
[2][35][2,73]. Therefore, there is an urgent unmet medical need for an agent that can deliver meaningful clinical efficacy in APOE4/4 homozygotes, without the increased risk of brain edema or microhemorrhage.
Since the oral anti-amyloid oligomer agent ALZ-801/valiltramiprosate inhibits the formation of Aβ oligomers without affecting plaques
[37][59], ALZ-801 has the potential to be a suitable therapeutic for APOE4 carriers that does not increase the risk of ARIA.
3. Interaction of Aβ and APOE4 with Other Molecules in AD Brain
The interaction of Aβ species with various proteins in the AD brain is an area of increasing importance, and studies of the Aβ interactome have the potential to provide novel therapeutic targets
[38][75]. One of the most studied protein interactions in AD is that of Aβ and APOE4
[39][40][76,77]. There are multiple mechanisms by which the APOE4 genotype confers increased AD risk, including decreased Aβ phagocytosis and clearance through the blood–brain barrier, increased tau hyperphosphorylation and aggregation, and exaggerated microglial and astrocytic responses. These effects collectively lead to diminished Aβ clearance, exaggerated neuroinflammation, and increased Aβ aggregation
[41][42][78,79], all leading to increased levels of neurotoxic soluble Aβ oligomers and tau spreading in the brains of APOE4 carriers, especially APOE4/4 homozygotes
[43][56]. Consistent with having the greatest amyloid burden and tau pathology, APOE4/4 subjects exhibit an earlier and faster rate of cognitive decline, becoming symptomatic approximately a decade earlier than non-carriers.
The interaction of APOE4 with the microglial activating receptor TREM2 is an emerging area of focus for drug discovery
[44][45][80,81]. This is a complex relationship, with the effects of microglial activation being homeostatic and protective in the early stages of AD and pro-inflammatory and harmful at later stages. Therefore, TREM2-targeted drug development is challenging since the benefits of TREM2 activation may be specific to the disease stage.
Recent studies have highlighted the APOE4 interaction with the Reelin-Disabled-1 (Dab1) signaling pathway, which plays a protective role in synaptic development and plasticity
[46][82]. Reelin signaling protects synapses from Aβ-induced neuronal stress, while APOE4 interferes with this protective effect, worsening synaptic toxicity of Aβ
[47][83]. Genetic variations in the Reelin pathway have been described in a Spanish population to confer increased risk
[48][84] and in the UK Biobank as risk factors for AD, particularly in APOE4/4 homozygotes
[49][85]. A recent report described a protective Reelin variant in a male with autosomal-dominant AD
[50][86].
4. Biomarkers and Biological Definition of Alzheimer’s Disease
The biological definition of AD has been essential for the success of drug trials with disease-modifying agents that target amyloid pathology, the only approach to date that has shown positive clinical and biomarker effects in delaying the progression of the disease. A clinical AD diagnosis without biomarker confirmation has very low accuracy in APOE4 non-carriers (~60%) and low accuracy in heterozygotes (80%) but has excellent accuracy in APOE4/4 homozygotes (>95%)
[51][87].
New insights into the pathogenic role of Aβ and the application of brain biomarkers have markedly improved our ability to identify individuals with AD pathology long before the onset of clinical symptoms and have led to diagnostic criteria for clinical research based on objective disease biomarkers
[12]. Data from longitudinal AD studies and interventional clinical trials using PET imaging of amyloid and tau aggregates and fluid biomarkers of AD were most helpful in advancing a biological definition of AD that avoids the pitfalls and substantial inaccuracy of reaching a clinical diagnosis, which was 30% in older clinical trials
[12][51][12,87]. The onset of biomarker changes closely correlates with clinical onset and stages of AD and follows a well-defined sequence of pathological changes. An increase in brain Aβ concentration leads to amyloid accumulation into toxic soluble oligomers and protofibrils, initiating tau phosphorylation that can be detected by amyloid and tau PET scans. An increase in CSF or plasma biomarkers, including Aβ, p-tau, and neurofilament light chain protein (NfL), precedes brain volume loss that can be assessed by brain volumetric MRI and ultimately results in cognitive decline
[52][53][54][55][88,89,90,91].
Biomarker-based AD diagnostic criteria hold the potential to enable the detection of pathological changes years and decades before the onset of clinical symptoms and require that subjects have positive amyloid and tau biomarkers, with or without evidence of neuronal injury or neurodegeneration. A/T/N classification is based on the status of amyloid (A), tau (T), and neuronal pathology (N), as determined by amyloid and tau PET imaging or CSF or plasma biomarkers, including Aβ, p-tau-, NfL, and neurogranin, and by MRI measurements of brain volume loss focused on hippocampal volume and cortical thickness.
P-tau has recently emerged as the most reliable diagnostic and staging marker in AD. Tau, a cytoskeletal protein that forms the scaffolding of neurons called microtubules, is phosphorylated at threonine 181 or 217 sites when neurons are stressed and injured by toxic amyloid oligomers
[52][53][56][46,88,89]. Longitudinal studies in AD subjects have shown that as levels of aggregated forms of amyloid increase in the brain, they induce abnormal phosphorylation of neuronal tau and a progressive elevation of p-tau in the CSF and plasma. These progressive increases in CSF and plasma p-tau precede the appearance of intraneuronal neurofibrillary tangles, the tau pathology that can be detected by tau PET imaging
[54][90]. An increase in the concentration of toxic amyloid oligomers induces downstream synaptic dysfunction and neuronal injury, leading to the formation of p-tau, as shown in
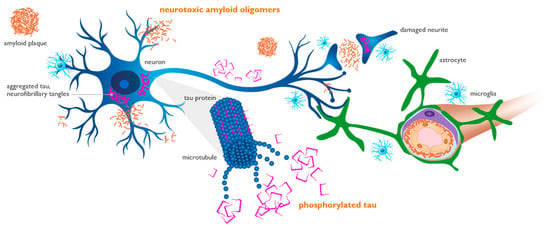
Figure 3.
Figure 3. Amyloid oligomers induce synaptic dysfunction and neuronal injury, leading to the phosphorylation of tau protein. Amyloid oligomers injure synapses located on neuronal dendrites and cell bodies (blue cell). Phosphorylated tau (p-tau) is released from injured neurons into the interstitial fluid and can be measured in the cerebrospinal fluid and plasma, representing neuronal dysfunction and loss. Aggregated tau (purple) is shown as twisted neurofibrillary tangles within neurons, forming tau tangles. Perivascular astrocytes (green cell) play an active role in the trafficking and clearance of beta-amyloid (Aβ) and p-tau through the glymphatic perivascular system into systemic circulation, where they can be detected by plasma assays.
P-tau
181 and p-tau
217 isoforms are found in the brain, CSF, and plasma of patients with AD pathology decades before clinical onset and have been shown to appear as a response to neurotoxic soluble amyloid aggregates
[54][55][90,91]. These p-tau isoforms are specific to AD and serve as reliable biomarkers for tracking disease progression and for determining the impact of effective anti-amyloid therapeutics
[52][53][55][88,89,91].
CSF assays for core AD biomarkers have been continually optimized, and some are approved for clinical use. More recently, plasma biomarker assays have also become sensitive and reliable for use in clinical trials. The late-stage amyloid antibody trials have included the evaluation of plasma biomarkers
[3][4][57][58][3,4,94,95], focusing on p-tau
181 and p-tau
217 isoforms, which have been associated with efficacy on standard clinical endpoints.
Anti-amyloid antibodies with meaningful clinical efficacy show plasma p-tau
181 reductions of ≥15% from baseline over 1 year, while those that failed to achieve efficacy reduced p-tau
181 by <10% over 1 year. This suggests that (1) the magnitude of plasma p-tau
181 reduction over 1 year is a reasonable predictor of clinical efficacy and (2) early and sustained reduction in p-tau
181 may be a suitable marker of target engagement and meaningful clinical efficacy. Lecanemab also showed a reduction in the synaptic injury marker neurogranin in the CSF, whereas NfL levels in the CSF did not separate from the placebo over 78 weeks of treatment. Both lecanemab and donanemab showed a significant reduction in the astrocytic marker plasma glial fibrillary acidic protein (GFAP) over 78 weeks
[4][57][59][4,92,94].
The dose of oral ALZ-801/valiltramiprosate being used in current clinical trials provides CNS concentrations that fully block the formation of neurotoxic soluble oligomers in mechanism of action studies
[60][37]. This dose has shown the most pronounced p-tau
181 reduction compared with the effects reported with other anti-amyloid agents, including anti-amyloid antibodies
[61][62][26,58]. This is consistent with the promising clinical efficacy that has been observed in APOE4 carriers treated with ALZ-801′s active agent tramiprosate.