New data suggest that the aggregation of misfolded native proteins initiates and drives the pathogenic cascade that leads to Alzheimer’s disease (AD) and other age-related neurodegenerative disorders. Researchers propose a unifying single toxin theory of brain neurodegeneration that identifies new targets and approaches to the development of disease-modifying treatments. An extensive body of genetic evidence suggests soluble aggregates of beta-amyloid (Aβ) as the primary neurotoxin in the pathogenesis of AD. New insights from fluid biomarkers, imaging, and clinical studies provide further evidence for the decisive impact of toxic Aβ species in the initiation and progression of AD.
- Alzheimer’s disease
- neurodegeneration
- disease modification
- beta-amyloid oligomers
- APOE4
- ALZ-801
- valiltramiprosate
- aducanumab
- lecanemab
- donanemab
1. Introduction
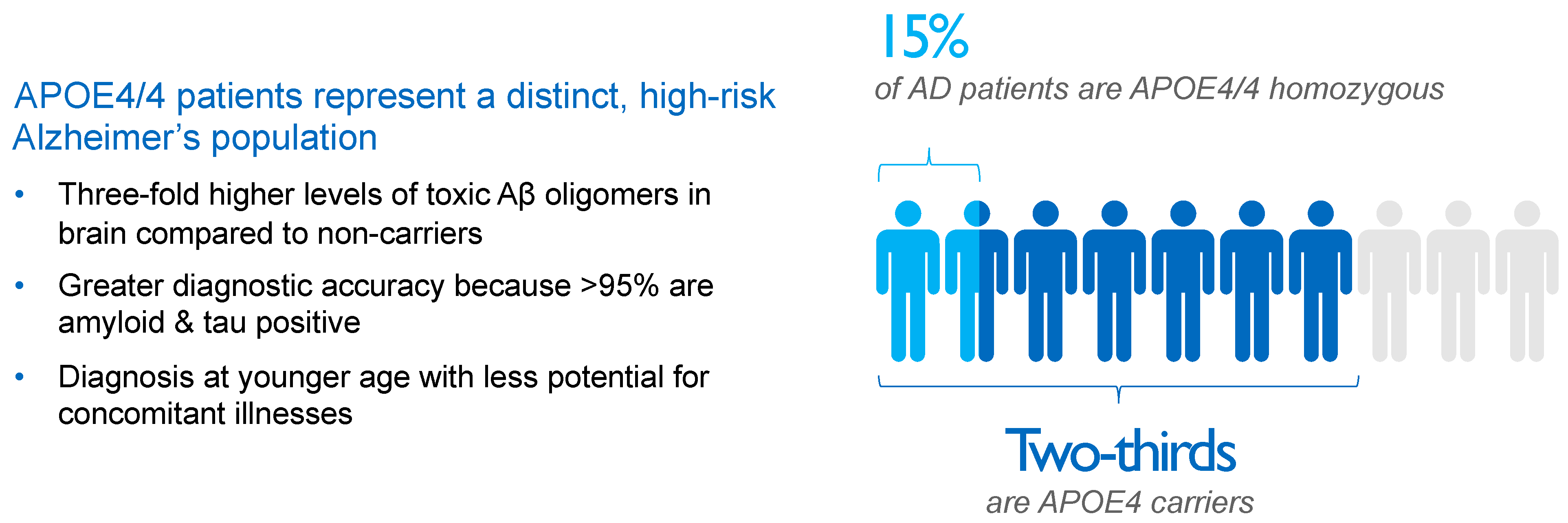
2. APOE4 Represents Main Genetic Risk Factor for Alzheimer’s Disease
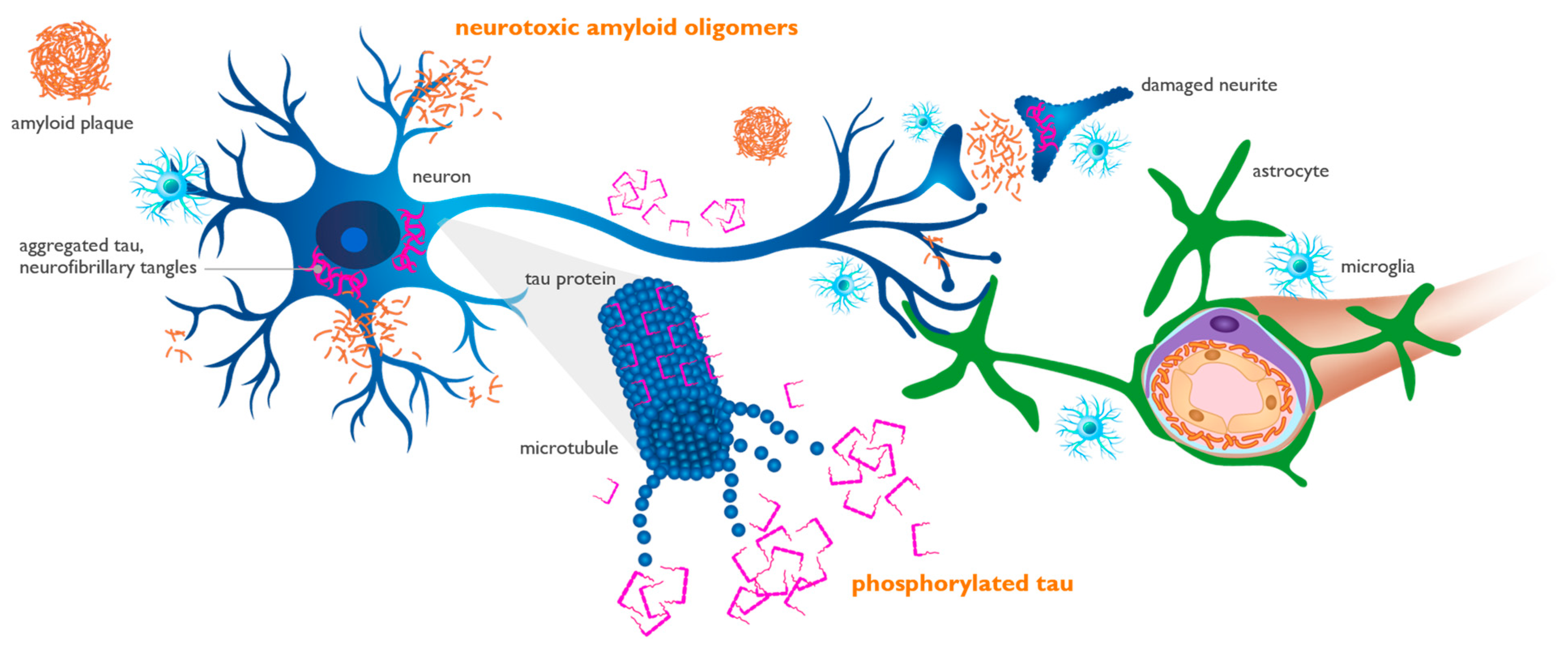
3. Interaction of Aβ and APOE4 with Other Molecules in AD Brain
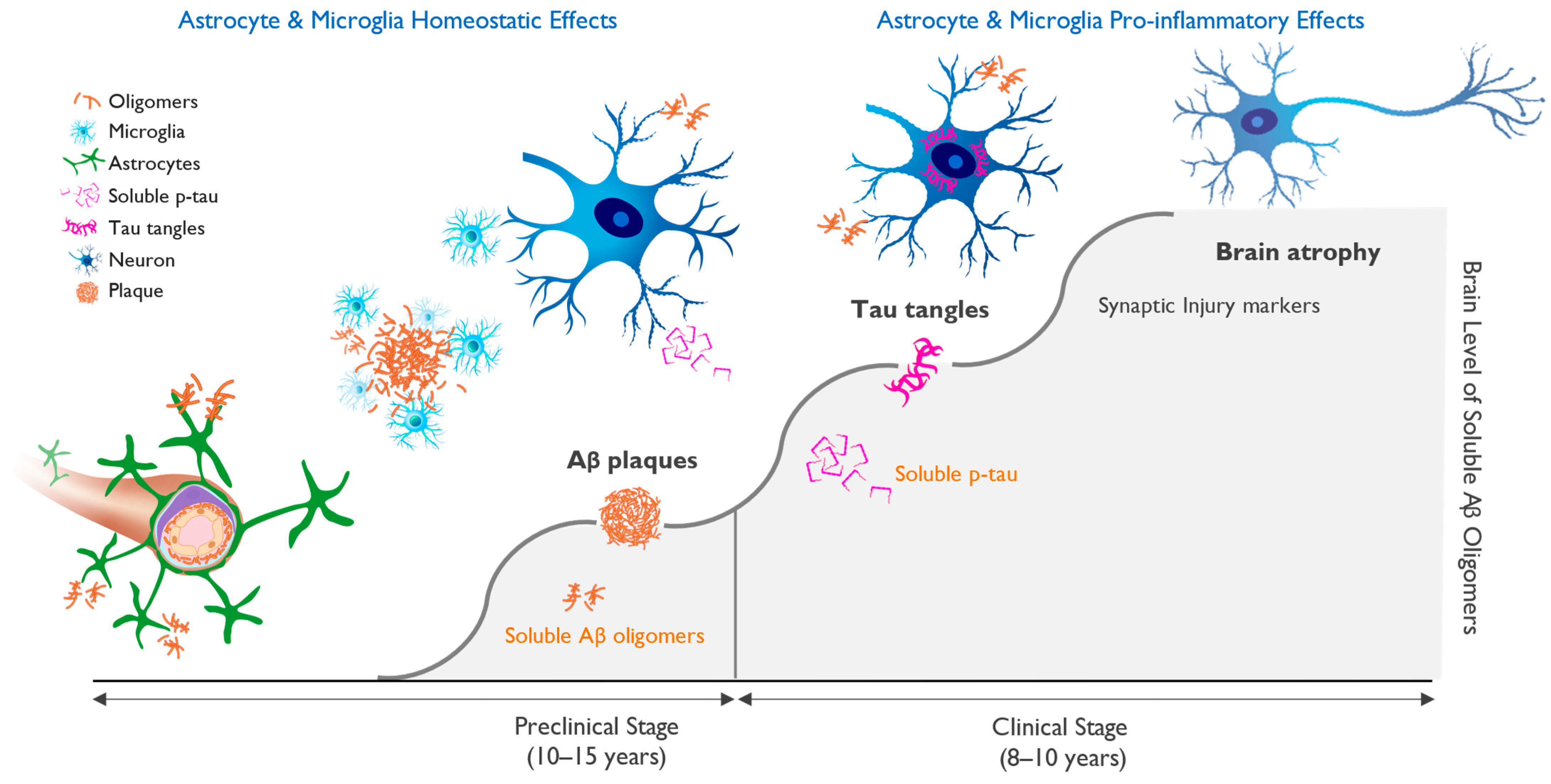
4. Biomarkers and Biological Definition of Alzheimer’s Disease
This entry is adapted from the peer-reviewed paper 10.3390/ijms25052727
References
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560.
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95.
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210.
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Eng. J. Med. 2023, 388, 9–21.
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in early symptomatic Alzheimer disease The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 2023, 330, 512–527.
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235.
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell. Longev. 2014, 2014, 795375.
- Hefter, D.; Draguhn, A. APP as a protective factor in acute neuronal insults. Front. Mol. Neurosci. 2017, 10, 22.
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133.
- Duran-Aniotz, C.; Moreno-Gonzalez, I.; Gamez, N.; Perez-Urrutia, N.; Vegas-Gomez, L.; Soto, C.; Morales, R. Amyloid pathology arrangements in Alzheimer’s disease brains modulate in vivo seeding capability. Acta Neuropathol. Commun. 2021, 9, 56.
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and a-synuclein in neurodegeneration. Brain 2017, 140, 266–278.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive Alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355.
- Wang, Z.; Weaver, D.F. Microglia and microglial-based receptors in the pathogenesis and treatment of Alzheimer’s disease. Int. Immunopharmacol. 2022, 110, 109070.
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 2020, 40, 1956–1974.
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wan, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimers Res. Ther. 2022, 14, 59.
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303.
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938.
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid PET Study Group; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949.
- Tijms, B.M.; Vromen, E.M.; Mjaavatten, O.; Holstege, H.; Reus, L.M.; van der Lee, S.; Wesenhagen, K.E.J.; Lorenzini, L.; Vermunt, L.; Venkatraghavan, V.; et al. Cerebrospinal fluid proteomics in patients with Alzheimer’s disease reveals five molecular subtypes with distinct genetic risk profiles. Nat. Aging 2024, 4, 33–47.
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Eng. J. Med. 2012, 367, 795–804.
- Rafii, M.S.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; Lott, I.; Klunk, W.; Head, E.; Christian, B.; et al. The AT(N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement. 2020, 12, e12062.
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997.
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: Correlation with amyloid PET. Brain 2017, 140, 1486–1498.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923.
- Saddiki, H.; Fayosse, A.; Cognat, E.; Sabia, S.; Engelborghs, S.; Wallon, D.; Alexopoulos, P.; Blennow, K.; Zetterberg, H.; Parnetti, L.; et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020, 17, e1003289.
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 13–14.
- Ward, A.; Crean, S.; Mercaldi, C.J.; Collins, J.M.; Boyd, D.; Cook, M.N.; Arrighi, H.M. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: A systematic review and meta-analysis. Neuroepidemiology 2012, 38, 1–17.
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653.
- Chalmers, K.; Wilcock, G.K.; Love, S. APOE ε4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol. Appl. Neurobiol. 2003, 29, 231–238.
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42.
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011, 7, 367–385.
- Withington, C.G.; Turner, R.S. Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer’s disease. Front. Neurol. 2022, 13, 862369.
- Leqembi (lecanemab-irmb) Injection for Intravenous Use ; Eisai Inc.: Nutley, NJ, USA, 2023; Available online: https://www.leqembi.com/ (accessed on 14 September 2023).
- Abushakra, S.; Mandelbaum, R.; Barakos, J.; Scheltens, P.; Porsteinsson, A.P.; Watson, D.; MacSweeney, E.; Sabbagh, M.; Liang, E.; Kesslak, P.; et al. Prevalence of amyloid-related imaging abnormalities in APOE4/4 homozygotes with early Alzheimer’s disease (AD): Baseline findings from ongoing clinical trials of the oral anti-amyloid agent ALZ-801 (valiltramiprosate) (P5-6.003). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023.
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: Integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs 2017, 31, 495–509.
- Lazarev, V.F.; Dutysheva, E.A.; Kanunikov, I.E.; Guzhova, I.V.; Margulis, B.A. Protein interactome of amyloid-β as a therapeutic target. Pharmaceuticals 2023, 16, 312.
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. J. Mol. Biol. 2019, 431, 2248–2265.
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754.
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192.
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7.
- Gratuze, M.; Schlachetzki, J.C.M.; D’Oliveira Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O’Brien, S.; et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 2023, 111, 202–219.e7.
- Jolly-Amado, A.; Kulkarni, N.; Nash, K.R. Reelin signaling in neurodevelopmental disorders and neurodegenerative diseases. Brain Sci. 2023, 13, 1479.
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid β toxicity. Sci. Signal. 2015, 8, ra67.
- Bufill, E.; Roura-Poch, P.; Sala-Matavera, I.; Antón, S.; Lleó, A.; Sánchez-Saudinós, B.; Tomàs-Abadal, L.; Puig, T.; Abós, J.; Bernades, S.; et al. Reelin signaling pathway genotypes and Alzheimer disease in a Spanish population. Alzheimer Dis. Assoc. Disord. 2015, 29, 169–172.
- Bracher-Smith, M.; Leonenko, G.; Baker, E.; Crawford, K.; Graham, A.C.; Salih, D.A.; Howell, B.W.; Hardy, J.; Escott-Price, V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2022, 119, 67–76.
- Lopera, F.; Marino, C.; Chandrahas, A.S.; O’Hare, M.; Villalba-Moreno, N.D.; Aguillon, D.; Baena, A.; Sanchez, J.S.; Vila-Castelar, C.; Ramirez Gomez, L.; et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 2023, 29, 1243–1252.
- Degenhardt, E.K.; Witte, M.M.; Case, M.G.; Yu, P.; Henley, D.B.; Hochstetler, H.M.; D’Souza, D.N.; Trzepacz, P.T. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 2016, 57, 208–216.
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433.
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018, 14, 1470–1481.
- Mattsson-Calgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz2387.
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 2022, 28, 1797–1801.
- Blennow, K.; Zetterberg, H. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog. Mol. Biol. Transl. Sci. 2019, 168, 3–23.
- Pontecorvo, M.J.; Lu, M.; Burnham, S.C.; Schade, A.E.; Dage, J.L.; Shcherbinin, S.; Collins, E.C.; Sims, J.R.; Mintun, M.A. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: A secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022, 79, 1250–1259.
- Bittner, T.; Blennow, K.; Scelsi, M.; Palermo, G.; Kollmorgen, G.; Smith, J.; Zetterberg, H.; Doody, R.S. GRADUATE I and II Results. Effect of Subcutaneous Gantenerumab on Fluid Biomarkers of AD Pathology and Neurodegeneration with Insights on Plasma Biomarkers and New CSF Biomarkers. Available online: https://medically.gene.com/global/en/unrestricted/neuroscience/adpd-2023/adpd-2023-presentation-bittner-graduate-i-and-ii-result.html (accessed on 14 September 2023).
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; de Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta (1–42/1–40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res. Ther. 2020, 12, 118.
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs 2018, 32, 849–861.
- Abushakra, S.; Hey, J.; Blennow, K.; Scheltens, P.; Reiman, E.M.; Hort, J.; Sheardova, K.; Rutgers, S.M.; Prins, N.D.; Dautzenberg, P.; et al. Effects of oral ALZ-801, an amyloid oligomer inhibitor, on plasma biomarkers in APOE4 carriers with Early Alzheimer’s disease: Results of six-month interim analysis from a phase 2 biomarker study. Alzheimers Dement. 2022, 18, e069141. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/epdf/10.1002/alz.069141 (accessed on 14 September 2023).
- Hey, J.H.; Abushakra, S.; Blennow, K.; Scheltens, P.; Hort, J.; Sheardova, K.; Prins, N.D.; Rutgers, M.S.; Dautzenberg, P.L.; Pazdert, L.; et al. Effects of ALZ-801, an oral amyloid oligomer inhibitor, on biomarkers of Alzheimer’s disease (AD): 12-month results of phase 2 biomarker study in early AD (S26.0007). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023; Available online: https://www.aan.com/MSA/Public/Events/AbstractDetails/52624 (accessed on 14 September 2023).