 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ali Mohammed Ali | -- | 1896 | 2024-01-31 08:56:08 | | | |
| 2 | Wendy Huang | Meta information modification | 1896 | 2024-01-31 11:12:23 | | |
Video Upload Options
Chronic progressive external ophthalmoplegia (CPEO) is the most common manifestation of mitochondrial diseases and is characterized by bilateral symmetrical progressive ptosis and reduced ocular motility. CPEO can be isolated or accompanied by a clinical feature of systemic involvement of mitochondrial dysfunction (CPEO plus syndrome). Mitochondrial disorders generally affect tissues with high metabolic demand, such as the central and peripheral nervous systems, heart, adrenal glands, renal tubules, skeletal muscles, and the eye. In CPEO, the ocular findings of ptosis and ophthalmoplegia occur due to the inability of the abnormal mitochondria to supply an adequate amount of ATP due to defective oxidative phosphorylation. The extraocular muscles are particularly susceptible due to their high mitochondrial volume and lower mutational threshold. Their susceptibility is expressed in multiple mitochondrial disorders, highlighting the significance of examining other manifestations in patients with PEO.
1. Introduction
2. CPEO
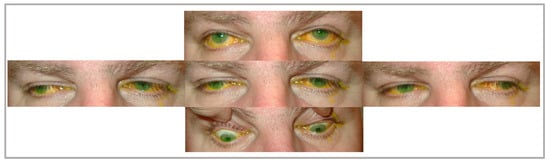
3. Kearns–Sayre Syndrome
4. Pearson Syndrome
5. Leigh Syndrome
6. MELAS
References
- Yu-Wai-Man, P.; Clements Al Nesbitt, V.; Griffiths, P.G.; Gorman, G.S.; Schaefer, A.M.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. A national epidemiological study of chronic progressive external ophthalmoplegia in the United Kingdom—Molecular genetic features and neurological burden. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5109.
- Kearns, T.P.; Sayre, G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA Arch. Ophthalmol. 1958, 60, 280–289.
- Olson, W.; Engel, W.K.; Walsh, G.O.; Einaugler, R. Oculocraniosomatic neuromuscular disease with “ragged-red” fibers. Arch. Neurol. 1972, 26, 193–211.
- Zintz, R.; Villiger, W. Electron microscopic findings in 3 cases of chronic progressive ocular muscular dystrophy. Ophthalmologica 1967, 153, 439–459.
- Reske-Nielsen, E.; Lou, H.C. Lowes, MProgressive external ophthalmoplegia. Evidence for a generalised mitochondrial disease with a defect in pyruvate metabolism. Acta Ophthalmol. 1976, 54, 553–573.
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719.
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311.
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000, 289, 782–785.
- Thangaraj, K.; Khan, N.A.; Govindaraj, P.; Meena, A.K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J. Med. Res. 2015, 141, 13–26.
- Chatzistefanou, K.I.; Brouzas, D.; Asproudis, I.; Tsina, E.; Droutsas, K.D.; Koutsandrea, C. Strabismus surgery for diplopia in chronic progressive external ophthalmoplegia. Int. Ophthalmol. 2019, 39, 213–217.
- Richardson, C.; Smith, T.; Schaefer, A.; Turnbull, D.; Griffiths, P. Ocular motility findings in chronic progressive external ophthalmoplegia. Eye 2005, 19, 258–263.
- Pfeffer, G.; Sirrs, S.; Wade, N.K.; Mezei, M.M. Multisystem Disorder in Late-Onset Chronic Progressive External Ophthalmoplegia. Can. J. Neurol. Sci. 2011, 38, 119–123.
- Sun, M.G.; Rojdamrongratana, D.; Rosenblatt, M.I.; Aakalu, V.K.; Yu, C.Q. 3D printing for low cost, rapid prototyping of eyelid crutches. Orbit 2019, 38, 342–346.
- Ahn, J.; Kim, N.J.; Choung, H.K.; Hwang, S.W.; Sung, M.; Lee, M.J.; Khwarg, S.I. Frontalis sling operation using silicone rod for the correction of ptosis in chronic progressive external ophthalmoplegia. Br. J. Ophthalmol. 2008, 92, 1685–1688.
- Rajabi, M.T.; Tabatabaie, S.Z.; Rajabi, M.B.; Abrishami, Y.; Hosseini, S.S.; Oestreicher, J. Management of myogenic ptosis in chronic progressive external ophtalmoplegia. Iran. J. Neurol. 2014, 13, 185–187.
- Shemesh, A.; Margolin, E. Kearns Sayre Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Ortiz, A.; Arias, J.; Cárdenas, P.; Villamil, J.; Peralta, M.; Escaf, L.C.; Ortiz, J. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. Int. J. Retin. Vitr. 2017, 3, 24.
- Kang, Y.X.; Wang, Y.J.; Zhang, Q.; Pang, X.H.; Gu, W. A case of hypopituitarism accompanying Kearns–Sayre syndrome treated with human chorionic gonadotropin: A case report and literature review. Andrologia 2017, 49, e12711.
- Ng, Y.S.; Lim, A.Z.; Panagiotou, G.; Turnbull, D.M.; Walker, M. Endocrine Manifestations and New Developments in Mitochondrial Disease. Endocr. Rev. 2021, 43, 583–609.
- Katsanos, K.H.; Nastos, D.; Noussias, V.; Christodoulou, D.; Kappas, A.; Tsianos, E.V. Manometric study in Kearns–Sayre syndrome. Dis. Esophagus 2001, 14, 63–66.
- Vadhul, R.; Halbach, C.S.; Areaux, R.G., Jr.; Berry, S.; Hou, J.H. Endothelial dysfunction in a child with Pearson marrow-pancreas syndrome managed with Descemet stripping automated endothelial keratoplasty using a suture pull-through technique. Digit. J. Ophthalmol. 2019, 25, 59–64.
- Shoeleh, C.; Donato, U.M.; Galligan, A.; Vitko, J. A Case Report on Pearson Syndrome with Emphasis on Genetic Screening in Patients Presenting with Sideroblastic Anemia and Lactic Acidosis. Cureus 2023, 15, e33963.
- Jennifer, M.S.; Cortez, D. Pearson marrow-pancreas syndrome with cardiac conduction abnormality necessitating prophylactic pacemaker implantation. Ann. Noninvasive Electrocardiol. 2020, 25, e12681.
- Reddy, J.; Jose, J.; Prakash, A.; Devi, S. Pearson syndrome: A rare inborn error of metabolism with bone marrow morphology providing a clue to diagnosis. Sudan. J. Paediatr. 2019, 19, 161–164.
- Broomfield, A.; Sweeney, M.G.; Woodward, C.E.; Fratter, C.; Morris, A.M.; Leonard, J.V.; Abulhoul, L.; Grunewald, S.; Clayton, P.T.; Hanna, M.G.; et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015, 38, 445–457.
- Faraci, M.; Cuzzubbo, D.; Micalizzi, C.; Lanino, E.; Morreale, G.; Dallorso, S.; Castagnola, E.; Schiaffino, M.C.; Bruno, C.; Rossi, A.; et al. Allogeneic bone marrow transplantation for Pearson’s syndrome. Bone Marrow Transplant. 2007, 39, 563–565.
- Saini, A.G.; Chatterjee, D.; Bhagwat, C.; Vyas, S.; Attri, S.V. Leigh syndrome in an infant: Autopsy and histopathology findings. Autops. Case Rep. 2021, 11, e2021334.
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203.
- Kistol, D.; Tsygankova, P.; Krylova, T.; Bychkov, I.; Itkis, Y.; Nikolaeva, E.; Mikhailova, S.; Sumina, M.; Pechatnikova, N.; Kurbatov, S.; et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023, 24, 1597.
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634.
- Han, J.; Lee, Y.-M.; Kim, S.M.; Han, S.Y.; Lee, J.B. Ophthalmological manifestations in patients with Leigh syndrome. Br. J. Ophthalmol. 2015, 99, 528–535.
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lönnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018, 55, 21–27.
- Brecht, M.; Richardson, M.; Taranath, A.; Grist, S.; Thorburn, D.; Bratkovic, D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015, 19, 95–100.
- Ardissone, A.; Bruno, C.; Diodato, D.; Donati, A.; Ghezzi, D.; Lamantea, E.; Lamperti, C.; Mancuso, M.; Martinelli, D.; Primiano, G.; et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: A study from the Italian network of mitochondrial diseases. Orphanet J. Rare Dis. 2021, 16, 413.
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Gonçalves, F.G.; Russo, F.L.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232.
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265.
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017, 40, 685–693.
- Pek, N.M.Q.; Phua, Q.H.; Ho, B.X.; Pang, J.K.S.; Hor, J.-H.; An, O.; Yang, H.H.; Yu, Y.; Fan, Y.; Ng, S.-Y.; et al. Mitochondrial 3243A>G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019, 10, 802.
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.; Battista, V.; Koenigsberger, D.; Pascual, J.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology 2011, 77, 1965–1971.
- Seed, L.M.; Dean, A.; Krishnakumar, D.; Phyu, P.; Horvath, R.; Harijan, P.D. Molecular and neurological features of MELAS syndrome in paediatric patients: A case series and review of the literature. Mol. Genet. Genom. Med. 2022, 10, e1955.
- Al-Enezi, M.; Al-Saleh, H.; Nasser, M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 2008, 15, 81–86.
- Weiduschat, N.; Kaufmann, P.; Mao, X.; Engelstad, K.M.; Hinton, V.; DiMauro, S.; De Vivo, D.; Shungu, D. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology 2014, 82, 798–805.
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012, 107, 247–252.
- Yoshida, T.; Ouchi, A.; Miura, D.; Shimoji, K.; Kinjo, K.; Sueyoshi, T.; Jonosono, M.; Rajput, V. MELAS and reversible vasoconstriction of the major cerebral arteries. Intern. Med. 2013, 52, 1389–1392.
- Cheng, W.; Zhang, Y.; He, L. MRI Features of Stroke-Like Episodes in Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-Like Episodes. Front. Neurol. 2022, 13, 843386.
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Horvath, R.; Klopstock, T.; Mancuso, M.; Martikainen, M.H.; Mcfarland, R.; Nesbitt, V.; Pitceathly, R.D.S.; et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019, 4, 201.
- Argudo, J.M.; Moncayo, O.M.A.; Insuasti, W.; Garofalo, G.; Aguirre, A.S.; Encalada, S.; Villamarin, J.; Oña, S.; Tenemaza, M.G.; Eissa-Garcés, A.; et al. Arginine for the Treatment of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes: A Systematic Review. Cureus 2022, 14, e32709.
- Pérez-Cruz, E.; González-Rivera, C.; Valencia-Olvera, L.d.C.G. Immunonutrition for the acute treatment of MELAS syndrome. Endocrinología. Diabetes Nutr. 2022, 69, 144–148.
- Li, J.; Zhang, W.; Cui, Z.; Li, Z.; Jiang, T.; Meng, H. Epilepsy Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front. Neurol. 2021, 12, 675816.

