2. CPEO
In its isolated form, CPEO is typically a sporadic disorder characterized by progressive bilateral ptosis and ophthalmoparesis [
9] (
Figure 1). Ptosis examination yields poor levator palpebrae superioris (LPS) muscle function, where eyelid excursion is often less than 8–10 mm rather than the normal ≥12 mm. Slowed, incomplete, and omnidirectional saccades can be a subtle early clinical sign that is frequently missed. Later on, as the disease progresses, ophthalmoplegia becomes more evident. The often-symmetric nature of the disease means that patients do not have diplopia, and reports of manifest strabismus with diplopia in CPEO patients are rare [
14,
15]. Retinal examination could reveal pigmentary retinopathy that is typical in Kearns–Sayre syndrome, characterized as salt and pepper retinopathy, where clumps of retinal pigment epithelium (RPE) alternate with areas devoid of RPE [
16]. However, these retinal changes rarely harbor field defects or a change in visual acuity.
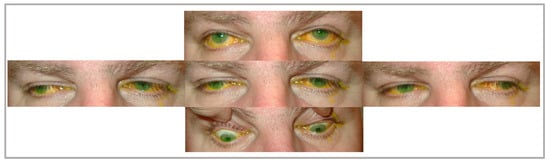
Figure 1. Patient A, diagnosed with mitochondrial encephalomyopathy, presents with chronic progressive external ophthalmoplegia with limited eye movements in all gazes and cerebellar signs (intention tremor in finger-to-nose test and tandem walking), in addition to areas of pigment hyperplasia on fundoscopy. The yellow discoloration shown in the image is from fluorescein eye staining.
Treatment of CPEO is focused on the correction of ptosis. It can start with eyelid crutches as a non-surgical solution, which usually is not preferred due to discomfort or intolerable aesthetics [
17]. Surgery is the mainstay treatment and is dependent on LPS function. Resection of the levator tendon along the superior tarsus is available for mild LPS impairment, while in more severe cases, frontalis suspension procedures with facia lata or silicon are used [
18,
19]. When strabismus and diplopia occur, prismatic glasses are prescribed to correct small malalignments, and strabismus surgery can be performed to improve the patient’s quality of life [
19].
3. Kearns–Sayre Syndrome
Kearns–Sayre syndrome is a syndrome of CPEO and pigmentary retinopathy, with onset before the age of 20 as well as one of the following features: a complete heart block, cerebellar ataxia, dementia, deafness, short stature, endocrine abnormalities, and cerebrospinal fluid (CSF) protein of more than 100 mg/dL. If the diagnostic criteria are not met, the patient is termed “CPEO plus” or “KSS-minus” [
20].
When a patient presents with CPEO before the age of 20, they should be evaluated with mtDNA sequencing followed by regular ophthalmologic assessments and screening for systemic signs and symptoms. A muscle biopsy can be performed to look for the ragged red fibers. The fundoscopic examination reveals pigmentary retinopathy that should be distinguished from retinitis pigmentosa since they might share similar symptoms like mildly reduced night vision and visual acuity. Retinitis pigmentosa typically affects the peripheral or the mid-peripheral retina with a bone spicule pattern, whereas KSS affects the posterior retina with a salt and pepper pattern [
21]. It is essential to perform an electrocardiogram on these patients to rule out a complete heart block. Endocrine abnormalities affecting the adrenals, parathyroid, and hypothalamus can present with diabetes mellitus, growth hormone deficiency, and short stature [
22,
23]. Orbicularis oculi muscle weakness can impair eyelid closure, and frontalis weakness can affect eyelid elevation. Dysphagia is a rare presentation of KSS and may result from upper esophageal sphincter dysfunction and reduced peristalsis in the pharynx and upper esophagus, as observed in a manometric study of a case report by Shaker et al. [
24].
No definitive treatment option is available for KSS. Symptomatic treatment includes correction of CPEO, treating heart blocks with pacemakers with a long-term cardiology follow-up, correction of endocrine abnormalities, and cochlear implants in cases of hearing loss.
4. Pearson Syndrome
Pearson syndrome (PS), also known as Pearson marrow–pancreas syndrome, is a rare fatal multisystemic mitochondrial disease due to deletions in mtDNA, and it typically affects infants. Ophthalmologic manifestations include corneal endothelial dysfunction, ptosis, CPEO, and mild peripheral pigmentary retinopathy [
25]. It is also characterized by refractory sideroblastic anemia, lactic acidosis, and exocrine pancreatic dysfunction. It can also present with vacuolization of hematopoietic precursors, pancytopenia, failure to thrive, diarrhea, hypospadias, cleft lip palate, diabetes mellitus, renal tubular dysfunction, hepatic failure, enteropathy, and rashes [
26]. Cardiac manifestations, such as bundle branch blocks and supraventricular tachycardia, have been reported; however, cardiac involvement is not yet a part of the major criterion of the disease [
27].
Usually, premature death at three years of age occurs due to infection from neutropenia or metabolic crisis. Thus, early diagnosis is essential in improving the poor prognosis for these patients. The diagnosis of Pearson syndrome is challenging due to the atypical presentation in infancy. It can be confirmed via mtDNA sequencing and observing multiple deletions of varying lengths [
28]. Interestingly, these single large-scale mtDNA deletions can also be found in young patients with CPEO and KSS. They, therefore, form a continuous spectrum of diseases termed “mtDNA deletion syndromes”, supported by reports of a KSS-like phenotype in PS survivors [
29].
Treatment for Pearson syndrome is supportive and may include blood transfusions, iron chelating therapy, pancreatic replacement therapy, and prompt detection and management of cardiac dysfunction. Bone marrow transplant has been tested and, unfortunately, yielded poor outcomes [
26,
30].
5. Leigh Syndrome
Leigh syndrome is a fatal, progressive neurodegenerative disease that typically manifests in infants and young children of 3 months to 2 years of age [
31]. It can be caused by multiple mtDNA deletions as well as nDNA defects in more than 75 different monogenic causes, most commonly by the
SURF1 variant [
32,
33].
The clinical features of LS vary, with the most common characteristics, according to a meta-analysis by Chang et al., being developmental delay, hypotonia, respiratory dysfunction, epilepsy, reduced feeding, and weakness [
34]. The ocular features of LS include nystagmus, ptosis, ophthalmoplegia, strabismus, pigmentary retinopathy, and optic atrophy [
34,
35]. Common cardiac abnormalities are hypertrophic or dilated cardiomyopathy and conduction defects such as Wolff–Parkinson–White syndrome [
36,
37].
Consensus on the clinical diagnosis is yet to be determined; however, LS is suspected through the hallmarks of the disease along with findings suggestive of brainstem dysfunction in addition to T2 weighted brain MRI lesions and accessory laboratory findings [
34]. Brain MRI findings typically show bilateral symmetrical supra-tentorial (basal ganglia, thalamus, and sub-thalamus) and/or infra-tentorial (brainstem and dentate nuclei) lesions. A study by Ardissone et al. presented a predominating basal ganglia involvement of 90.2%. They also showed that both supra and infra-tentorial involvement is dominant in cases of both mtDNA (74%) and -nDNA (67%) variants, while isolated infra-tentorial variants are rare [
38]. Extensive research is being conducted to find genetic correlations with MRI findings of LS. For example, a retrospective cohort found significant associations between the
SURF1 variant and inferior olivary nuclei lesions [
39].
Abnormal laboratory findings may yield elevated blood, urine, and CSF lactate levels. Additional deficiencies may be observed in respiratory chain complexes through enzyme assays and pyruvate dehydrogenase complex [
40]. However, these laboratory findings are not consistently present. Therefore, confirmatory tests with genetic assays are required for a definitive diagnosis and the identification of specific variants of LS [
41].
6. MELAS
MELAS, or mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (SLEs) are associated with A to G RNA transfer mutation (Leu (UUR)) in the most commonly
m.3243A>G mutation [
42,
43].
The clinical presentations vary widely, usually in childhood, with neurological symptoms that include SLEs, sensorineural hearing loss, and cognitive impairment associated with diffuse white matter injury. Less commonly, it can present with gastrointestinal manifestations that include gastric perforation, ischemic colitis, segmental ileal paralysis, pseudo-obstruction, or megacolon. Endocrine manifestations, such as diabetes mellitus, have also been reported in MELAS [
44].
Ophthalmologic manifestations of MELAS include hemianopia and cortical blindness from SLEs, nystagmus, cataracts, CPEO, optic atrophy, salt and pepper pigmentary retinopathy, and macular degeneration [
45].
The transient SLEs of the disease are characterized by nausea, vomiting, a migraine-like headache, encephalopathy, and focal seizures with or without neurological deficits. The exact pathogenic mechanism for these episodes is yet to be determined; however, three theories have been postulated. The first is insufficient energy due to mitochondrial dysfunction, supported by the increase in lactate peaks and decreased N-acetyl aspartate peaks of the occipital regions in brain magnetic resonance spectroscopy (MRS) [
46]. The second is nitric oxide (NO) deficiency, which usually regulates oxygenation and blood flow. This hypothesis is supported by a reduction in NO metabolites during acute attacks and an increase in NO synthase inhibitors in the COX-negative fibers of MELAS patients [
47]. The third theory is mitochondrial angiopathy, an accumulation of mitochondria in the smooth muscle cells and endothelial cells of small cerebral arteries leading to the narrowing of the lumen of blood vessels and reducing perfusion [
48]. MRI findings of SLE exhibit stroke-like lesions (SLLs) that are usually differentiated from other pathologies by initially observing cortical and deep white matter lesions, in addition to occipital and parietal lobe lesions or lesions not confined to arterial territories. PWI/ASL can also show hyperperfused lesions, and MRS exhibits lactate peaks [
49]. Another distinctive finding in neuroimaging was reported in some cases of MELAS as cerebellar lesions SLLs [
49,
50].
Since MELAS is associated with reduced levels of citrulline and arginine, which are NO precursors, and decreased NO that contributes to SLEs, supplement replacement with arginine was proposed. A systematic review by Argudo et al. concluded that the studies conducted showed promising results in managing SLEs [
51]. Acute phase management consists of giving an intravenous dose of 500 mg/kg/day or 10 g/m
2 in 24 h for 3–5 days. Whereas chronically, 150–300 mg/kg/day (maximum of 500 mg) is used instead [
52]. A study conducted by Pek et al. using induced pluripotent stem cell-derived endothelial cells vouched for edaravone, a potent antioxidant, to be used for improving the vascular function in MELAS since it scavenges ROS and inhibits the inflammatory response in cerebrovascular diseases, which L-arginine and citrulline do not tackle [
42]. For treating epilepsy, levetiracetam is considered to be the first-line anticonvulsant in mitochondrial encephalomyopathy due to the mitochondrial toxicity of other anticonvulsant agents [
53].