Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diego Ruano | -- | 2383 | 2024-01-23 10:42:15 | | | |
| 2 | Jessie Wu | + 8 word(s) | 2391 | 2024-01-24 02:05:23 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gavilán, E.; Medina-Guzman, R.; Bahatyrevich-Kharitonik, B.; Ruano, D. Protein Quality Control Systems in SARS-CoV-2 Infection. Encyclopedia. Available online: https://encyclopedia.pub/entry/54244 (accessed on 10 August 2026).
Gavilán E, Medina-Guzman R, Bahatyrevich-Kharitonik B, Ruano D. Protein Quality Control Systems in SARS-CoV-2 Infection. Encyclopedia. Available at: https://encyclopedia.pub/entry/54244. Accessed August 10, 2026.
Gavilán, Elena, Rafael Medina-Guzman, Bazhena Bahatyrevich-Kharitonik, Diego Ruano. "Protein Quality Control Systems in SARS-CoV-2 Infection" Encyclopedia, https://encyclopedia.pub/entry/54244 (accessed August 10, 2026).
Gavilán, E., Medina-Guzman, R., Bahatyrevich-Kharitonik, B., & Ruano, D. (2024, January 23). Protein Quality Control Systems in SARS-CoV-2 Infection. In Encyclopedia. https://encyclopedia.pub/entry/54244
Gavilán, Elena, et al. "Protein Quality Control Systems in SARS-CoV-2 Infection." Encyclopedia. Web. 23 January, 2024.
Copy Citation
SARS-CoV-2’s structure and mechanism of infection have been well characterized. The virus comprises a lipid envelope studded with spike (S) proteins. These spikes facilitate viral entry into host cells by binding to angiotensin-converting enzyme 2 (ACE2) receptors on the cell surface. Following attachment, the virus enters the cell by endocytosis. Its genetic material consists of a single-stranded RNA molecule, which encodes structural proteins, non-structural proteins (NSP), and accessory proteins. Once inside, the viral RNA is translated into proteins, including those for replication and the formation of new virus particles.
COVID-19
SARS-CoV-2
protein quality control systems
ER stress
neurodegeneration
1. Endoplasmic Reticulum Stress and Unfolded Protein Response
Endoplasmic reticulum (ER) stress and unfolded protein response (UPR) are common aspects that emerge during the appearance and progression of both neurodegenerative diseases [1][2][3] and viral infections [4][5]. Viral infections often trigger an upsurge in protein synthesis, potentially surpassing the folding capacity of the ER. Consequently, this imbalance leads to the accumulation of unfolded proteins, inducing ER stress [4][5]. In response to ER stress, cells activate a multifaceted signaling network known as UPR, an adaptive response aimed at mitigating the burden of unfolded proteins to sustain cellular viability and function [6][7].
The UPR is a sophisticated signaling pathway initiated by the activation of three primary UPR stress sensors (see Figure 1): inositol-requiring protein 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [4][6][8][9][10]. The glucose regulated protein-78 (GRP78, also known as BiP or HSPA5), is an ER chaperone that plays a pivotal role as the principal regulatory protein by binding to these sensors. In the absence of stress, GRP78 predominantly associates with these three proteins, effectively suppressing their activity and preventing the initiation of UPR signaling. However, under stress conditions characterized by the accumulation of misfolded proteins in the ER (as observed in neurodegenerative diseases and in viral infections), GRP78 interacts with these unfolded proteins, aiming to maintain their foldable state and resulting in the release of the three UPR mediators (Figure 1). The specific mechanisms of the UPR pathways include [8][9]:
- −
-
IRE1 activation and splicing of XBP1 (X-box binding protein 1) mRNA, resulting in the production of sXBP1, an active transcription factor. sXBP1 regulates the expression of chaperones and ERAD components, reinforcing ER’s protein-folding and -degradation capacity. IRE1 also catalyzes the degradation of a large number of mRNAs and some pre-microRNAs (pre-miRNAs). This process is called regulated IRE1-dependent decay (RIDD) [11][12]. On the other hand, it is known that IRE1 is capable of forming high-order complexes in the ER membrane and interacting with a large number of proteins, among which the tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) stands out. This interaction activates a cascade of signaling that leads to the activation of c-Jun N-terminal kinase (JNK), which, in turn, can inhibit some anti-apoptotic members of the BCL-2 family while activating pro-apoptotic proteins. Together, these two events lead to the oligomerization of BCL-2-like protein 4 (BAX) and BCL-2 antagonist/killer (BAK), initiating the apoptosis process [11][12][13];
- −
-
PERK activation and the phosphorylation of the eukaryotic initiation factor 2α (eIF2α), a pivotal regulator of protein translation. Phosphorylated eIF2α reduces global protein synthesis, thereby alleviating the ER burden and allowing cells to cope with ER stress. However, this process can also induce apoptosis by upregulating the C/EBP homologous protein (CHOP). The prolonged upregulation of CHOP induces apoptosis through pathways involving the BCL2 binding component 3 (BBC3/PUMA) and the tribbles pseudokinase 3 (TRIB3).
- −
-
ATF6 activation and translocation to the Golgi apparatus, where it is cleaved by site-1 (S1P) and site-2 (S2P) proteases, releasing a 50 kDa N-terminal fragment that translocates to the nucleus. This fragment acts as a transcription factor that subsequently upregulates genes encoding ER chaperones and other proteins involved in ER quality control.
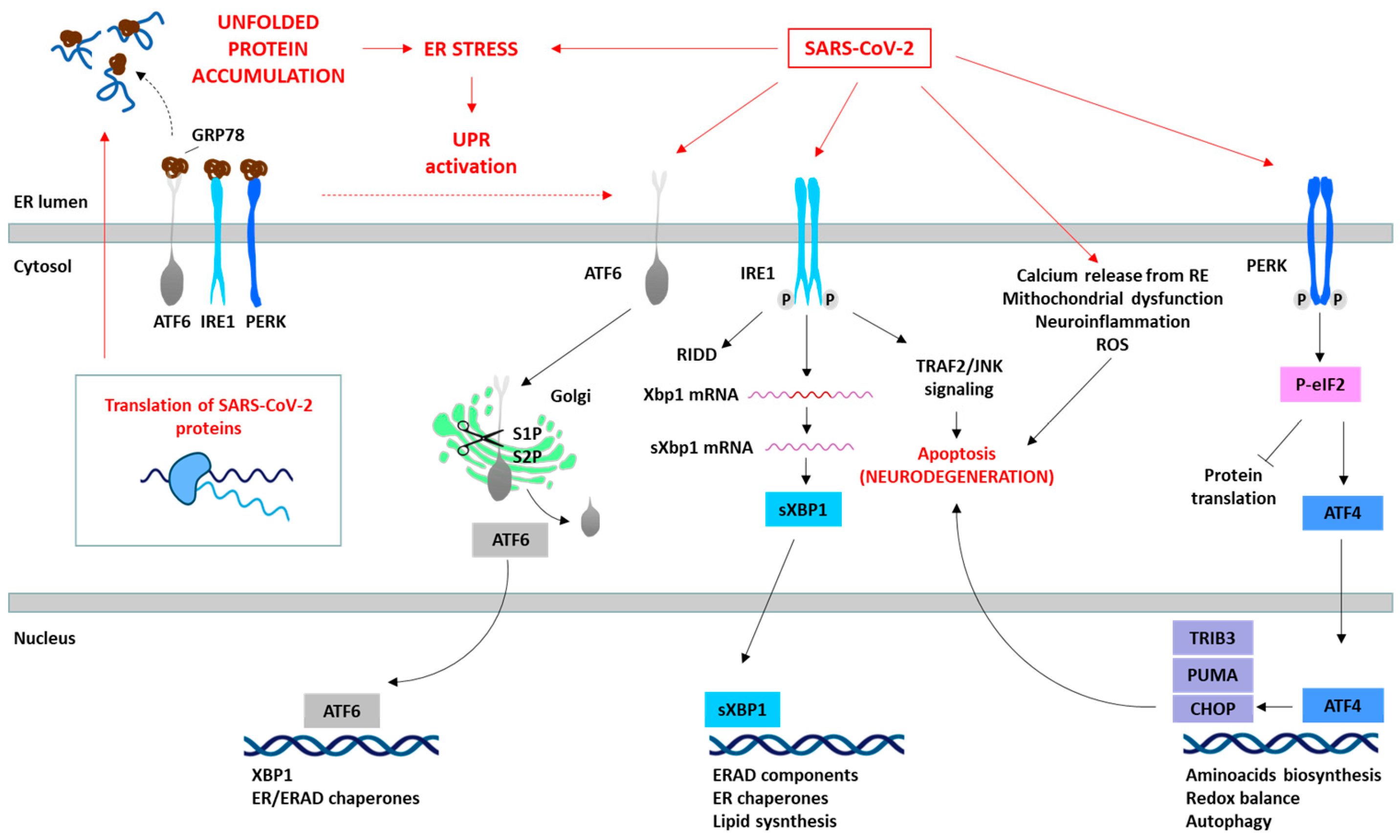
Figure 1. SARS-CoV-2 induces ER stress and activates the UPR. The translation of SARS-CoV-2 proteins generates ER stress, which activates the UPR as an antiviral host cell response. However, SARS-CoV-2 is able to manipulate the different pathways of the UPR (red arrows) depending on the viral requirements for its replication. The specific proteins involved in the SARS-CoV-2 and UPR interaction are summarized. The prolonged activation of the UPR leads to the initiation of pro-apoptotic signaling pathways (e.g., IRE1/TRAF2/JNK or PERK/ATF4/CHOP), disturbances in cellular calcium levels due to ER release, mitochondrial dysfunction, and consequently ROS generation, and the induction of oxidative stress, culminating in cell death.
These sensors orchestrate adaptive processes through both transcriptional and non-transcriptional responses to restore ER homeostasis (Figure 1). This involves adapting protein synthesis, enhancing protein folding capacity, and increasing the efficiency of ERAD [8][9][10]. The activation of these three UPR pathways leads to diverse downstream consequences, depending on the nature and intensity of the stimuli and the specific cell type involved. Although the primary objective of the UPR is to reinstate ER homeostasis, prolonged or severe ER stress can activate apoptotic pathways through factors such as activating transcription factor 4 (ATF4) and CHOP, ultimately culminating in cell death, as observed in neurodegenerative processes [4][8][9][10].
2. Endoplasmic Reticulum-Associated Degradation and Ubiquitin-Proteasome System
ERAD is a crucial process in the quality control of ER proteins, facilitating the elimination of aberrant proteins by the UPS [14]. This process is essential for maintaining cellular proteostasis, as misfolded or unassembled proteins can compromise the cell function. This process consists of three phases: recognition of misfolded or damaged proteins in the ER, retrotranslocation into the cytosol, and ubiquitin-dependent degradation by the proteasome (Figure 2). ER luminal chaperones both prevent improper folding and identify terminally misfolded proteins, directing them for ERAD. Some ERAD components include the EDEM family members, such as EDEM1, EDEM2, and EDEM3, as well as the Osteosarcoma 9 (OS9) protein, the chaperone GRP78, and protein disulfide-isomerases (PDIs), such as the ER DnaJ domain-containing protein 5 (ERdj5). These components facilitate the recognition, recruitment and disposal of the misfolded proteins [15][16] for retrotranslocation via the ERAD complex. This complex involves the Sec61 translocon, DERL1-3 and valosin-containing protein (p97), together with the suppressor enhancer Lin12 1-like (SEL1L) and the ERAD-associated E3 ubiquitin–protein ligase (HRD1) that contribute to the ubiquitination process [16][17]. After (poly)ubiquitination, the proteins are recognized and degraded by the proteasome (Figure 1).
The ubiquitination process (which can take place in the ER or in the cytosol) involves the covalent attachment of ubiquitin molecules to lysine (K) residues on the substrate protein, through an enzymatic cascade involving E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin ligase) enzymes. The specificity of ubiquitination is determined by the type of linkage formed between ubiquitin molecules [18]. Hence, the E3 ubiquitin ligases play a pivotal role in specifying the target proteins for degradation. Their activity, expression, and turnover are rigorously regulated to prevent inappropriate ubiquitination and maintain cellular function [14]. The type of ubiquitination will direct the protein to a specific destination. For example, K48 linkages are tagged to UPS degradation, while K63 is tagged to autophagy and non-proteolytic signaling pathways [18][19].
The proteasome, a large multi-catalytic protease, is responsible for the degradation of ERAD substrates, as well as other UPS-tagged proteins [20]. The proteasome consists of a catalytic core complex (20S proteasome) and regulatory subunits such as 19S or 11S particles. The catalytic core is the 20S proteasome, a hollow barrel-shaped structure comprising four rings with α and β subunits. The α-rings control substrate access, and the β-rings house catalytic subunits (β1, β2, and β5). This 20S proteasome degrades non-ubiquitinated misfolded or damaged proteins [4]. Additionally, the proteasome can associate with regulatory subunits, forming different types of proteasomes (mainly 26S or 30S, immunoproteasome, hybrid proteasomes) with distinct functions. The regulatory particle 19S forms the 26S or 30S proteasome when associated with the 20S proteasome. The 19S particle facilitates binding, deubiquitination, unfolding, and the channeling of target proteins for degradation [4][20]. Additionally, deubiquitinating enzymes (DUBs) can modulate ubiquitination and the ERAD process by removing ubiquitin chains from substrate proteins [21]. Besides this, cytokines such as interferon α (IFNα), IFNγ and TNFα induce the replacement of constitutive catalytic subunits (β1, β2, and β5) in the 20S proteasome with inducible subunits β1i, β2i, and β5i, and this results in the formation of the immunoproteasome (Figure 2). The immunoproteasome, expressed in immune cells, exhibits distinct proteolytic activities compared to the standard 20S proteasome. It plays essential roles in antigen presentation, γ-interferon-mediated microglial activation, cytokine production by microglial cells, and the regulation of T-cell populations, highlighting its significance in immune responses and cellular regulation [22].
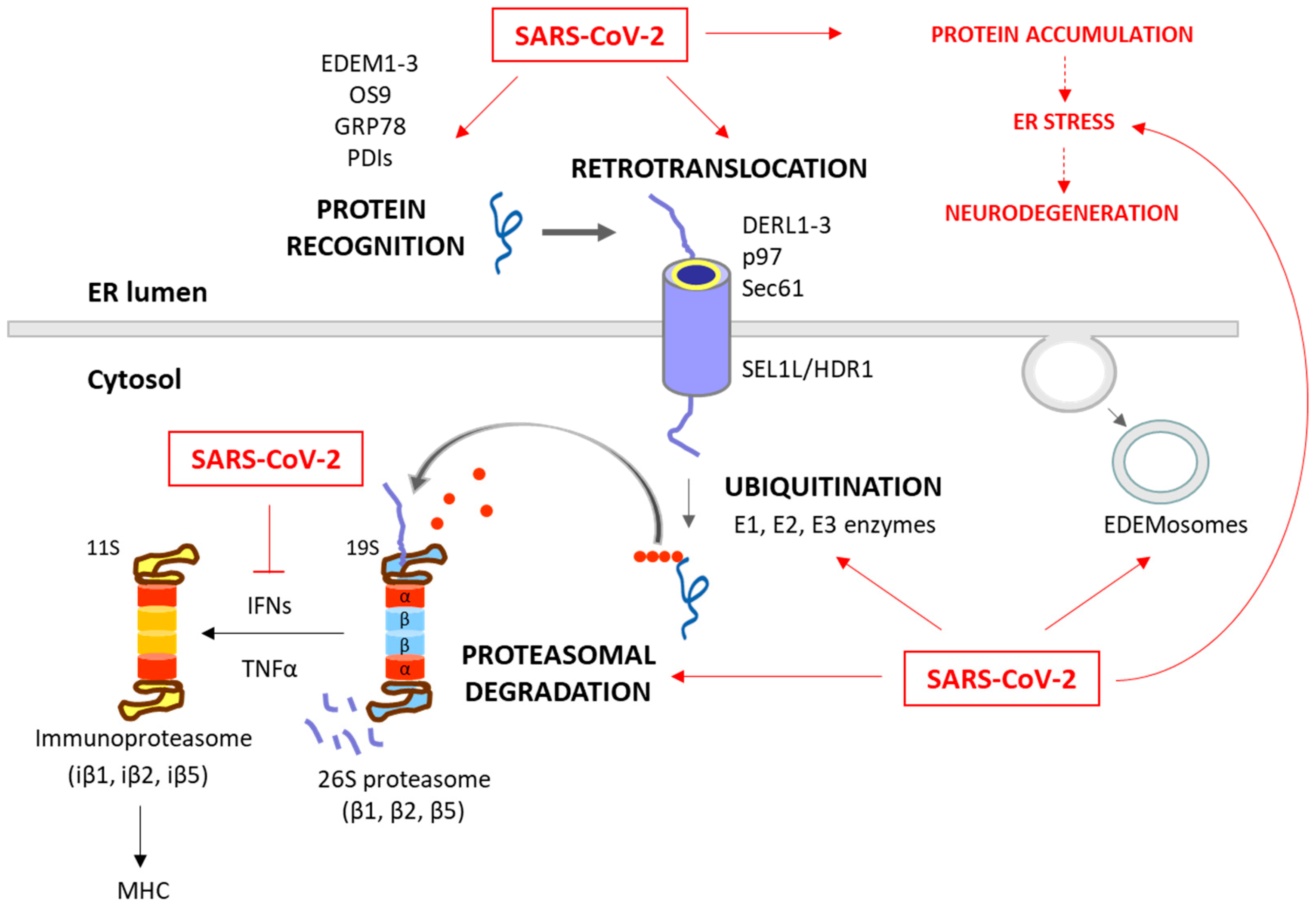
Figure 2. Degradation of unfolded protein through the ERAD/UPS pathways and SARS-CoV-2 interplay. When chaperones are not able to achieve the proper folding of proteins within the ER, these misfolded proteins are recognized and isolated by lectins and ERAD components, including EDEM1-3, OS9, PDIs and GRP78. The targeted proteins are then recruited to the ERAD complex and retrotranslocated to the cytosol. This complex involves Sec61, DERL1-3 and p97, together with SEL1L and HRD1, which assist in the ubiquitination process. After (poly)ubiquitination, the proteins are recognized by the 26S proteasome and deubiquitinated for degradation. While these processes are common antiviral responses of host cells, SARS-CoV-2 is able to positively or negatively manipulate this machinery to ensure its replication, escape and survival (red arrows). Consequently, the ERAD and UPS systems are not efficient in solving protein accumulation, leading to neurodegeneration. Moreover, SARS-CoV-2 can inhibit IFN production, and therefore the formation of immunoproteasome and antigen presentation.
The ERAD is also modulated by the ERAD tuning process, the regulation of ERAD activity by segregating ERAD components (like EDEM proteins) into specific ER-derived vesicles, named EDEMosomes (Figure 2). This segregation prevents the premature degradation of certain proteins, allowing the cell to regulate ERAD more precisely [23][24]. A huge number of associations between the ERAD pathway and human diseases have been established, highlighting the former’s significance in health and disease. Neurodegenerative diseases are characterized by disruptions in both ERAD and the UPS. These disruptions contribute to the accumulation of abnormal proteins and the formation of toxic protein aggregates, which are commonly associated with the pathogenesis of these conditions [25][26][27]. Also, beyond their fundamental roles in cell proteostasis, the ERAD and UPS have emerged as critical players in the intricate interaction between viruses and host cells during the process of viral infection [28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46].
3. Autophagy
Autophagy is a fundamental cellular process responsible for the degradation and recycling of cellular components, playing a crucial role in maintaining cellular homeostasis by removing damaged organelles and cellular debris, protein aggregates and specific soluble proteins [47][48]. There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). All of them culminate in the delivery of the target substrate to the lysosome for degradation. Macroautophagy involves the formation of autophagosomes that engulf cytoplasmic material, which then fuse with lysosomes [49]. Microautophagy directly engulfs cargo at the lysosomal membrane [50], while CMA selectively targets proteins recognized by specific chaperone proteins [51]. Each type of autophagy is orchestrated by a distinct set of key proteins [48]. In macroautophagy, initiation involves the contribution of unc-51-like autophagy-activating kinases 1 and 2 (ULK1/2), autophagy-relate gen 13 (ATG13), FAK family-interacting protein of 200 kDa (FIP200), and ATG101, while autophagosome nucleation relies on Beclin-1 (BECN1), vacuolar protein sorting 34 (VPS34), VPS15, and ATG14. Elongation requires ATG5, the ATG12–ATG5–ATG16L1 complex, ATG7, ATG10, and ATG3. The microtubule-associated protein 1A/1B-light chain 3 (LC3), the gamma-aminobutyric acid receptor-associated protein (GABARAP), the lysosomal-associated membrane protein 2 (LAMP2) and the sequestosome-1 (SQSTM1/p62, hereafter referred to as p62) participate in the recognition of cargo, and autophagosome maturation and fusion [49][52]. Microautophagy involves the endosomal sorting complexes required for transport (ESCRT) machinery and heat-shock cognate 70 (HSC70) in endosomal microautophagy, and LAMP2A and HSC70 in lysosomal microautophagy [50][53]. Chaperone-mediated autophagy relies on HSC70, co-chaperones, and LAMP2A, wherein KFERQ-like motifs guide substrate recognition and translocation into lysosomes [51][54]. These key proteins, together with other involved proteins, collectively govern the dynamic and selective degradation of cellular components, illustrating the intricate molecular machinery underlying the diverse autophagic pathways. Additionally, various specialized forms of autophagy have been defined, such as mitophagy (targeting damaged mitochondria), pexophagy (clearing peroxisomes), aggrephagy (removing protein aggregates), lipophagy (degrading lipid droplets), ER-phagy (eliminating parts of the ER), and nucleophagy (selective removal of nuclear components). Each targets specific cellular structures, ensuring the maintenance of cellular health through controlled degradation and recycling.
Autophagy plays a complex role in cellular health. Proper autophagic function is essential for neuronal survival, as it clears misfolded proteins associated with neurodegenerative diseases. However, dysfunctional autophagy can contribute to the accumulation of toxic protein aggregates, a hallmark of conditions like AD and PD disease, among other neurodegenerative diseases [55][56][57]. Numerous preclinical studies provide evidence supporting the potential use of autophagy modulators as suppressors of age-related pathologies, particularly in the context of neurodegenerative diseases [57]. In viral infections, similar to the ERAD/UPS systems, autophagy plays a dual role. On one hand, it serves as a crucial host defense mechanism by targeting and degrading viral components, contributing to the elimination of the virus. On the other hand, certain viruses have evolved to exploit the autophagic process for their own replication and survival, subverting the host defense mechanisms for their benefit [58][59][60]. This intricate interplay between autophagy and viral infections highlights the complexity of host–virus interactions and the multifaceted nature of cellular responses to viral invasion. As for neurodegeneration, modulating autophagic activity has emerged as a promising approach to combat viral infections [61].
4. Molecular Chaperones
Molecular chaperones are crucial cellular components for the maintenance of proteostasis. They are responsible for assisting in the proper folding of proteins, preventing misfolding, and aiding in the assembly and transport of proteins to their functional destinations [62]. These chaperones are localized in different cellular compartments, reflecting their diverse roles in assisting protein folding within specific environments. The major cellular locations for molecular chaperones include the cytoplasm (such as the heat shock proteins (HSPs) HSP70, HSP90 and Chaperonin/HSP60 families), ER (GRP78, GRP94, calnexin and calreticulin), mitochondria (HSP70 and HSP60), and the nucleus (HSP70 and HSP90) [63]. Their functional roles extend beyond their involvement in normal cellular functions; they play a critical role in various pathological conditions. In the context of neurodegenerative diseases, such as AD and PD, the misfolding and aggregation of specific proteins like tau and α-synuclein are associated with chaperone dysfunction [64][65][66]. Chaperones like HSP27, HSP70 and HSP90, which are typically involved in preventing protein aggregation, may become overwhelmed or less effective, contributing to the progression of these diseases [67][68]. On the other hand, viral infections can retain the host chaperones, prompting a kind of chaperone dysfunction due to the alterations in the chaperone activity and target substrates to be folded, induced by the virus [69][70][71]. Again, the viruses can perturb the protein quality control systems, probably facilitating the neurodegenerative process.
References
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127.
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491.
- Uddin, M.S.; Yu, W.S.; Lim, L.W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101417.
- Ruano, D. Proteostasis Dysfunction in Aged Mammalian Cells. The Stressful Role of Inflammation. Front. Mol. Biosci. 2021, 8, 658742.
- Cirone, M. ER Stress, UPR Activation and the Inflammatory Response to Viral Infection. Viruses 2021, 13, 798.
- Gavilan, M.P.; Pintado, C.; Gavilan, E.; Jimenez, S.; Rios, R.M.; Vitorica, J.; Castano, A.; Ruano, D. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 2009, 8, 654–665.
- Suaya, M.; Sanchez, G.M.; Vila, A.; Amante, A.; Cotarelo, M.; Garcia Carrillo, M.; Blaustein, M. Live and let die: Signaling AKTivation and UPRegulation dynamics in SARS-CoVs infection and cancer. Cell Death Dis. 2022, 13, 846.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102.
- Gomora-Garcia, J.C.; Geronimo-Olvera, C.; Perez-Martinez, X.; Massieu, L. IRE1α RIDD activity induced under ER stress drives neuronal death by the degradation of 14-3-3 θ mRNA in cortical neurons during glucose deprivation. Cell Death Discov. 2021, 7, 131.
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217.
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 2006, 312, 572–576.
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957.
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 2007, 581, 3658–3664.
- Morito, D.; Nagata, K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell 2015, 59, 335–344.
- Jeong, H.; Hong, E.H.; Ahn, J.H.; Cho, J.; Jeong, J.H.; Kim, C.W.; Yoon, B.I.; Koo, J.H.; Park, Y.Y.; Yang, Y.M.; et al. ERdj5 protects goblet cells from endoplasmic reticulum stress-mediated apoptosis under inflammatory conditions. Exp. Mol. Med. 2023, 55, 401–412.
- Lopata, A.; Kniss, A.; Lohr, F.; Rogov, V.V.; Dotsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369.
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Mol. Cell 2016, 64, 251–266.
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806.
- Trulsson, F.; Akimov, V.; Robu, M.; van Overbeek, N.; Berrocal, D.A.P.; Shah, R.G.; Cox, J.; Shah, G.M.; Blagoev, B.; Vertegaal, A.C.O. Deubiquitinating enzymes and the proteasome regulate preferential sets of ubiquitin substrates. Nat. Commun. 2022, 13, 2736.
- Paz Gavilan, M.; Vela, J.; Castano, A.; Ramos, B.; del Rio, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982.
- Movaqar, A.; Yaghoubi, A.; Rezaee, S.R.; Jamehdar, S.A.; Soleimanpour, S. Coronaviruses construct an interconnection way with ERAD and autophagy. Future Microbiol. 2021, 16, 1135–1151.
- Noack, J.; Bernasconi, R.; Molinari, M. How viruses hijack the ERAD tuning machinery. J. Virol. 2014, 88, 10272–10275.
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. Ubiquitin-Proteasome System in Neurodegenerative Disorders. J. Drug Metab. Toxicol. 2015, 6, 187.
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70.
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590.
- van Vliet, V.J.E.; Huynh, N.; Pala, J.; Patel, A.; Singer, A.; Slater, C.; Chung, J.; van Huizen, M.; Teyra, J.; Miersch, S.; et al. Ubiquitin variants potently inhibit SARS-CoV-2 PLpro and viral replication via a novel site distal to the protease active site. PLoS Pathog. 2022, 18, e1011065.
- Camborde, L.; Planchais, S.; Tournier, V.; Jakubiec, A.; Drugeon, G.; Lacassagne, E.; Pflieger, S.; Chenon, M.; Jupin, I. The ubiquitin-proteasome system regulates the accumulation of Turnip yellow mosaic virus RNA-dependent RNA polymerase during viral infection. Plant Cell 2010, 22, 3142–3152.
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021, 184, 3410–3425.e3417.
- Tang, Q.; Wu, P.; Chen, H.; Li, G. Pleiotropic roles of the ubiquitin-proteasome system during viral propagation. Life Sci. 2018, 207, 350–354.
- Kong, F.; You, H.; Kong, D.; Zheng, K.; Tang, R. The interaction of hepatitis B virus with the ubiquitin proteasome system in viral replication and associated pathogenesis. Virol. J. 2019, 16, 73.
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2013, 23, 85–96.
- Li, Z.; Hao, P.; Zhao, Z.; Gao, W.; Huan, C.; Li, L.; Chen, X.; Wang, H.; Jin, N.; Luo, Z.Q.; et al. The E3 ligase RNF5 restricts SARS-CoV-2 replication by targeting its envelope protein for degradation. Signal Transduct. Target. Ther. 2023, 8, 53.
- Chaudhary, P.; Proulx, J.; Park, I.W. Ubiquitin-protein ligase E3A (UBE3A) mediation of viral infection and human diseases. Virus Res. 2023, 335, 199191.
- Rojas, V.K.; Park, I.W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. Int. J. Mol. Sci. 2019, 20, 2984.
- Voss, M.; Braun, V.; Bredow, C.; Kloetzel, P.M.; Beling, A. Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication. Viruses 2021, 13, 1360.
- Zhao, M.; Zhang, M.; Yang, Z.; Zhou, Z.; Huang, J.; Zhao, B. Role of E3 ubiquitin ligases and deubiquitinating enzymes in SARS-CoV-2 infection. Front. Cell. Infect. Microbiol. 2023, 13, 1217383.
- Zu, S.; Li, C.; Li, L.; Deng, Y.Q.; Chen, X.; Luo, D.; Ye, Q.; Huang, Y.J.; Li, X.F.; Zhang, R.R.; et al. TRIM22 suppresses Zika virus replication by targeting NS1 and NS3 for proteasomal degradation. Cell Biosci. 2022, 12, 139.
- Le-Trilling, V.T.K.; Trilling, M. Ub to no good: How cytomegaloviruses exploit the ubiquitin proteasome system. Virus Res. 2020, 281, 197938.
- Han, K.; Zhao, D.; Liu, Y.; Liu, Q.; Huang, X.; Yang, J.; Zhang, L.; Li, Y. The ubiquitin-proteasome system is necessary for the replication of duck Tembusu virus. Microb. Pathog. 2019, 132, 362–368.
- Pang, Y.; Li, M.; Zhou, Y.; Liu, W.; Tao, R.; Zhang, H.; Xiao, S.; Fang, L. The ubiquitin proteasome system is necessary for efficient proliferation of porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2021, 253, 108947.
- Seyoum Tola, F. The Role of Ubiquitin-Proteasome System in the Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus-2 Disease. Int. J. Inflam. 2023, 2023, 6698069.
- Gao, W.; Wang, L.; Ju, X.; Zhao, S.; Li, Z.; Su, M.; Xu, J.; Wang, P.; Ding, Q.; Lv, G.; et al. The Deubiquitinase USP29 Promotes SARS-CoV-2 Virulence by Preventing Proteasome Degradation of ORF9b. mBio 2022, 13, e0130022.
- Ming, S.L.; Zhang, S.; Wang, Q.; Zeng, L.; Zhou, L.Y.; Wang, M.D.; Ma, Y.X.; Han, L.Q.; Zhong, K.; Zhu, H.S.; et al. Inhibition of USP14 influences alphaherpesvirus proliferation by degrading viral VP16 protein via ER stress-triggered selective autophagy. Autophagy 2022, 18, 1801–1821.
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral entry and the ubiquitin-proteasome system. Cell Microbiol. 2021, 23, e13276.
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830.
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Levine, B.; Kroemer, G. SnapShot: Macroautophagy. Cell 2008, 132, 162.e1–162.e3.
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136.
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39.
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41.
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203.
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Gavilan, E.; Pintado, C.; Gavilan, M.P.; Daza, P.; Sanchez-Aguayo, I.; Castano, A.; Ruano, D. Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Neurobiol. Aging 2015, 36, 1953–1963.
- He, C.; Klionsky, D.J. Autophagy and neurodegeneration. ACS Chem. Biol. 2006, 1, 211–213.
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650.
- Chen, T.; Tu, S.; Ding, L.; Jin, M.; Chen, H.; Zhou, H. The role of autophagy in viral infections. J. Biomed. Sci. 2023, 30, 5.
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78.
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354.
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730.
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332.
- Adriaenssens, E.; Asselbergh, B.; Rivera-Mejias, P.; Bervoets, S.; Vendredy, L.; De Winter, V.; Spaas, K.; de Rycke, R.; van Isterdael, G.; Impens, F.; et al. Small heat shock proteins operate as molecular chaperones in the mitochondrial intermembrane space. Nat. Cell Biol. 2023, 25, 467–480.
- Bobori, C.; Theocharopoulou, G.; Vlamos, P. Molecular Chaperones in Neurodegenerative Diseases: A Short Review. Adv. Exp. Med. Biol. 2017, 987, 219–231.
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes. Dev. 2008, 22, 1427–1438.
- Witt, S.N. Molecular chaperones, alpha-synuclein, and neurodegeneration. Mol. Neurobiol. 2013, 47, 552–560.
- Franklin, T.B.; Krueger-Naug, A.M.; Clarke, D.B.; Arrigo, A.P.; Currie, R.W. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int. J. Hyperth. 2005, 21, 379–392.
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976.
- Hooper, P.L.; Hightower, L.E.; Hooper, P.L. Loss of stress response as a consequence of viral infection: Implications for disease and therapy. Cell Stress Chaperones 2012, 17, 647–655.
- Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; Gammazza, A.M. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. J. Clin. Med. 2020, 9, 3518.
- Cheng, X.; Belshan, M.; Ratner, L. Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J. Virol. 2008, 82, 1229–1237.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
978
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
24 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No