Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alicja Nowaczyk | -- | 3887 | 2024-01-02 22:06:49 | | | |
| 2 | Lindsay Dong | Meta information modification | 3887 | 2024-01-03 02:52:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Stielow, M.; Witczyńska, A.; Kubryń, N.; Fijałkowski, �.; Nowaczyk, J.; Nowaczyk, A. The Bioavailability of Drugs. Encyclopedia. Available online: https://encyclopedia.pub/entry/53340 (accessed on 04 August 2026).
Stielow M, Witczyńska A, Kubryń N, Fijałkowski �, Nowaczyk J, Nowaczyk A. The Bioavailability of Drugs. Encyclopedia. Available at: https://encyclopedia.pub/entry/53340. Accessed August 04, 2026.
Stielow, Marlena, Adrianna Witczyńska, Natalia Kubryń, Łukasz Fijałkowski, Jacek Nowaczyk, Alicja Nowaczyk. "The Bioavailability of Drugs" Encyclopedia, https://encyclopedia.pub/entry/53340 (accessed August 04, 2026).
Stielow, M., Witczyńska, A., Kubryń, N., Fijałkowski, �., Nowaczyk, J., & Nowaczyk, A. (2024, January 02). The Bioavailability of Drugs. In Encyclopedia. https://encyclopedia.pub/entry/53340
Stielow, Marlena, et al. "The Bioavailability of Drugs." Encyclopedia. Web. 02 January, 2024.
Copy Citation
Drug bioavailability is a crucial aspect of pharmacology, affecting the effectiveness of drug therapy. Understanding how drugs are absorbed, distributed, metabolized, and eliminated in patients’ bodies is essential to ensure proper and safe treatment. In addition to biochemical activity, bioavailability also plays a critical role in achieving the desired therapeutic effects. This may seem obvious, but it is worth noting that a drug can only produce the expected effect if the proper level of concentration can be achieved at the desired point in a patient’s body. Given the differences between patients, drug dosages, and administration forms, understanding and controlling bioavailability has become a priority in pharmacology.
bioavailability
pharmacokinetics
therapeutic effectiveness
pharmaceutical innovations
1. Introduction
Drug bioavailability plays a crucial role in the effectiveness of pharmacological therapy. It determines the degree and rate at which drug-active substances are absorbed into the bloodstream after oral, topical, parenteral, and rectal administration [1]. In practice, bioavailability indicates the amount of the administered dose of a drug reaching the bloodstream in the form of the active ingredient which is then available to the body to produce a therapeutic effect [2].
Bioavailability is affected by variety of factors, including the medication’s physicochemical properties, the mode of administration, interactions with other substances, absorption, hepatic metabolism, and excretion [3][4]. The bioavailability of the active pharmaceutical ingredients (API) corresponds to the dose entering the bloodstream and consequently, its effectiveness [2]. For this reason, a drug’s bioavailability must be considered when designing a therapy and dosage. The efficient adjustment of an administered dosage requires knowledge of the API’s absorption, transport mechanism, metabolism, and elimination from the system [5]. Conversely, the ineffective administration of medications adds to the escalation of superfluous drug use. From this perspective, the significance of bioavailability becomes even more significant when it comes to patient safety and treatment efficacy [6]. Improving medication bioavailability can be accomplished by using suitable drug delivery methods, modifying drug formulations, optimizing dosages, identifying and controlling factors that decrease bioavailability, and monitoring blood drug levels resulting from dose adjustments. The monitoring of bioavailability can provide insights into drug interactions, as well as enable the development of tailored treatment plans for patients with liver or intestinal dysfunction [7]. Safe drug therapy requires good bioavailability. Insufficient bioavailability can reduce therapy efficacy, whereas excessive medication concentrations can produce toxicity and side effects [8]. Nevertheless, drug bioavailability is only one of many factors impacting the efficacy of drug therapy [9]. Currently, the field of pharmaceutical research and development is facing various challenges, one of which is the optimization of medication bioavailability. This optimization is pursued with the goal of enhancing the safety and efficacy of treatments [10].
2. Bioavailability of Drugs: Basic Concepts and Controlling Factors
The bioavailability of a drug describes the level of absorption of the active substance contained therein and the speed at which it is absorbed from the form administered to the patient, becoming available in the targeted location of the body, usually in the bloodstream. The bioavailability of a drug determines the proportion of the active substance available in the body in relation to its amount in the drug [11].
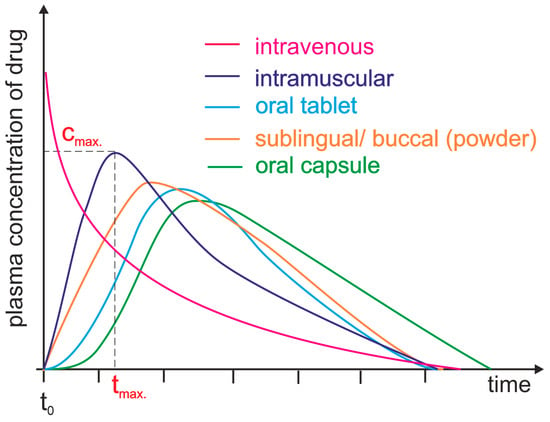
Drug bioavailability measurements allow for the assessment of the absorption efficiency of a drug, including its absolute and relative bioavailability, the time to reach maximum concentration, and the area under the concentration–time curve [12]. Absolute bioavailability is defined as a measurement that determines the percentage of the active substance entering the bloodstream after administration of a drug when the reference standard is an intravenous dose. It expresses the efficiency of the absorption of the active substance by the body. It is usually less than 100%, since not all of the active substance is absorbed in the gastrointestinal tract [13]. Relative bioavailability is expressed as the ratio of the bioavailability of two dosage forms of the same drug (Figure 1). The time to reach maximum concentration (tmax) measures the time it takes for an active ingredient to reach its highest concentration in the blood after drug administration. tmax is an important parameter because it can affect the rate of action of a drug. The area under the curve (AUC) measures the total amount of an active substance absorbed and available in the bloodstream as a function of time. The AUC is used to assess the total exposure of the body to an active substance (Figure 1) [14]. Measurements of drug bioavailability are essential for evaluating the pharmacokinetics and pharmacodynamics of drugs and for determining appropriate therapeutic doses. They are also crucial for developing new drugs and evaluating the efficacy of different administration forms [12][15].
Figure 1. Plasma level time curves for different types of drug administration. The drug is delivered directly into the systemic circulation via intravenous injection, ensuring 100% bioavailability and immediate achievement of maximum plasma concentration (cmax, tmax = 0 min). For other parenteral routes, such as subcutaneous and intramuscular injections, most drugs show between 60 and 100% bioavailability due to little or no metabolism in the skin or muscle. However, the time to reach maximum plasma concentration (tmax) is significantly longer than that achieved by intravenous administration. Orally administered drugs achieve a bioavailability level substantially lower than 100% due to incomplete absorption and/or elimination during the first pass through the liver. Additionally, due to the indirect path to the plasma, they are characterized by a long time lag. Different dosage forms may result in differences in cmax and tmax.
The ADME processes affect drug absorption, distribution, metabolism, and elimination in the body [16][17].
Absorption is the passing of a drug from the administration site (e.g., gastrointestinal tract, skin, respiratory system) into the bloodstream [18]. When administered orally, the drug must be absorbed from the gastrointestinal tract into the bloodstream to achieve its effect. Various factors can determine absorption, such as dosage form, the presence of food, the environment’s pH, or interactions with other substances [19].
Distribution is defined as the spread of an active compound throughout the body, leading to the presence of the drug in various tissues and organs after its absorption into the bloodstream. Distribution depends on several factors, such as vascular resistance, distribution volume, degree of drug binding to plasma proteins, and penetration of tissue barriers (e.g., permeability of the blood–brain barrier) [20]. Once a substance enters the body, part of it can bind to proteins in the blood, mainly albumin (acidic and neutral molecules, such as vitamins, drugs, and their metabolites), but also other proteins, such as acidic α1-glycoprotein and lipoproteins, as well as gamma globulins. Drug–protein complexes are too large to passively penetrate cell membranes, which affects drug distribution in the body. For the desired pharmacological effect to be achieved, most of the drug must be in free (unbound) form, since they can only interact with receptors in this form. If two or more drugs bind to the same plasma protein, they compete for a binding site, affecting their biological availability, as well as their effects [21].
The process by which the body, with the assistance of enzymes, transforms a drug into different substances—known as metabolites—is referred to as drug metabolism [22]. The liver is the most critical site of drug metabolism. This process aims to convert the drug into more soluble substances, which are more efficiently eliminated from the body, and to manage its concentration and activity [23].
Eliminating drugs from the body involves the removal of both the drug and its metabolites from the organism. The two main routes of elimination are excretion through urine and secretion via bile. The pharmacokinetic processes play a vital role in determining the appropriate drug doses, determining the optimal timing of administration, and assessing the therapeutic effectiveness. [14].
Factors influencing the bioavailability of drugs can be divided into four main categories: physicochemical agents, biological agents, pharmaceutical agents, and patient factors. The physicochemical properties of a drug are important for its bioavailability. Examples of physicochemical factors include drug solubility in the digestive environment, chemical stability, lipophilicity, ionizability, and pharmaceutical form.
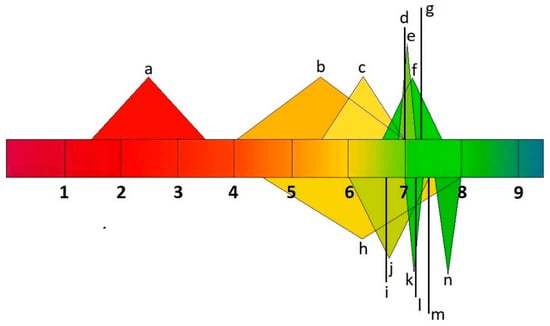
A drug must be soluble in the intestinal environment to be absorbed across biological membranes. Additionally, the ability of a drug to penetrate the membrane can be altered by its ionic form, which depends on the environment’s pH [24]. Drug molecules can ionize over different pH ranges, and the acid–base dissociation constant (pKa) is adopted as an universal measure of ionization. The pH range ϵ (2–12) indicates the point at which 63% of drugs ionize, according to the World Narcotics Index. About 43% and 12% of drugs, respectively, contain a single primary or acid center. The ionization process can significantly impact the properties of drug absorption, distribution, metabolism, excretion, and toxicity in vivo. Changing a drug’s ionic form (Figure 2) can affect its action, absorption, and therapeutic efficacy, depending on the environment’s pH [25][26].
Figure 2. pH values of human body fluids a: stomach; b: small intestine; c: large intestine; d: liver; e: muscle; f: uterus; g: testis; h: bladder; i: lungs; j: saliva; k: kidneys; l: brain, heart, and spleen; m: bone; n: pancreas.
The bioavailability of drugs can be significantly affected by various biological processes occurring in the body, such as gastric acidity, organ blood flow, digestive enzyme activity, intestinal microflora, and biological barriers. Gastric acidity, for example, may influence drug distribution and solubility. Furthermore, the activity of digestive enzymes in the gut can either reformulate the drug or reduce its availability, influencing its effectiveness [27]. Biological barriers, such as the intestinal cell membrane and blood–brain barrier, can limit the penetration of a drug into the bloodstream [28]. Biological barriers protecting the system against pathogens complicate drug delivery and distribution [29]. The adult brain possesses five barrier interfaces that regulate molecular movement into the brain parenchyma. These are the blood–brain barrier (BBB) the blood–cerebrospinal fluid barrier (BCSFB) [30], the blood–arachnoid barrier (BAB) [31], the circumventricular organs (CVOs) [32], and the ependyma [33]. The blood–brain barrier (BBB) is created by a tight structure of endothelial cells (ECs) joined together by protein couplings. These cells line the cerebral microvessels, separating the blood from the brain’s interstitial fluid [34]. The choroid plexus epithelium is between the blood and the ventricular cerebrospinal fluid (CSF) and forms the blood–CSF barrier. The epithelium between the blood and the subarachnoid CSF forms the arachnoid barriers. These three barrier layers limit and regulate molecular exchange at the interface between the blood and neural tissue or its fluid spaces. [35]. The inherent function of biological barriers impedes drug delivery and uptake, preventing effective therapeutic interventions. Biological barriers hinder treatment options and reduce the bioavailability of drugs in areas protected by barriers, which can ultimately lead to increased drug resistance [36].
Pharmaceutical factors encompass the technologies used to create a drug. These include drug formulation, excipients, formulation methods, and drug release techniques [37]. The main objective of any drug delivery system (DDS) is to maintain the desired therapeutic effect by providing and sustaining adequate drug concentration at the target site in the body. This involves enhancing drug efficacy; resolving issues related to solubility, low bioavailability, and poor in-body distribution; and minimizing side effects.
The drug formulation process often involves combining inactive ingredients and additional substances with APIs to produce drug products with specific characteristics. Improving this process to achieve an optimal drug formulation can involve various objectives such as increasing efficacy, extending the duration of therapeutic effects, reducing adverse effects, prolonging the shelf life of active ingredients, and enhancing compatibility with patient intake patterns [1]. APIs can be formulated using different material combinations, including neutral boosters such as polymers, lipids, surfactants, and other active ingredients, depending on the desired delivery method and specific application requirements. Such formulations are made possible by utilizing various types of delivery systems, including different kinds of microparticles (MPs), nanoparticles (NPs), and complex multi-component systems [2][3][4][5]. Typical practices involving these delivery mechanisms are often evolving, resulting in the development of drug products in various forms, such as solids, liquids, or non-oral administration methods [38].
Drug forms such as tablets, capsules, granules, powders, suspensions, solutions, emulsions, inserts, ointments, inserts, aerosols, patches, and transdermal systems affect the drug’s dissolution rate and absorption [39]. Excipients, such as binders, solvents, and stabilizers, can affect a drug’s bioavailability through interactions with the drug or changes in its solubility [40]. Drug release techniques can control the active ingredient’s release rate, which affects its availability and action [37]. Acetylsalicylic acid, commonly known as aspirin, is available in various administration forms. It can be taken orally as tablets in enteral, enteric-coated, effervescent, and controlled-release forms.
Patient factors refer to individual patient characteristics that can affect the bioavailability of a drug [41]. These include age, gender, genotype, health status, and diet. The drug absorption, metabolism, and elimination processes may differ depending on age. Children and the elderly may exhibit differences in metabolic enzymes, renal function, and blood flow, which can affect the bioavailability of drugs.
The protein known as P-glycoprotein (P-gp) plays a critical role in creating barriers within cells, particularly in the endothelial cells of the blood vessels. Its primary role is to prevent the entry of various substances, including drugs, into neural tissue by removing them from endothelial cells and returning them into the bloodstream. P-gp is a multidrug transporter that can recognize many compounds with different chemical structures and molecular weights (ranging from 330 to 4000 Da) [42]. It can transport hydrophobic and inert substances, as well as cations, but it cannot transport anions. The log p value of approximately 2.2 for DTG [43] indicates that it is only partially subject to bioaccumulation due to its moderate hydrophobicity [44]. P-gp is a protein crucial in transporting substances into and out of cells. Dolutegravir (DTG) is a substance for which P-gp is particularly important. Studies have shown that when DTG enters endothelial cells from the blood, it is pumped back into the bloodstream due to P-gp activity. However, disruption of the blood–brain barrier caused by HIV can lead to the dysfunction of P-gp, making it easier for drugs like DTG to penetrate brain tissues. This can result in higher concentrations of DTG in the brain, leading to unwanted side effects such as insomnia and headaches [45]. It is important to note that P-gp is present in tissues with a secretory function, such as the small intestine, liver, and kidney. If there is a pathological dysfunction of the P-gp protein, it can result in increased symptoms of dysfunction in these tissues. Recent studies reveal that P-gp triggers the production of effector T cells after viral infection.
3. Methods for Assessing Drug Bioavailability
Three main categories of methods are used to assess drug bioavailability—in vitro methods, in vivo methods, and new techniques and tools.
In vitro methods are laboratory-based and involve studying drug bioavailability under controlled conditions outside the living organism. These methods enable researchers to study drug absorption, metabolism, and transport processes and to evaluate the impact of physicochemical factors on drug bioavailability [46]. There are different in vitro methods used for drug testing. One such method is drug solubility testing, in which the solubility of a drug is measured in various environments, including the use of buffer solutions with different pH levels. This test helps determine how well a drug dissolves and is absorbed in the digestive environment [47].
Cell cultures are another example in which human or animal cells are used to simulate processes such as drug absorption, metabolism, and transport. These cultures can also be used to examine the effects of digestive enzyme activity or transporters on bioavailability [48]. Narrow liver microsomes containing microsomal enzymes accurately represent the liver’s metabolic activity, with various applications. Studying drug metabolism using hepatic microsomes allows for the estimation of how a drug may be metabolized before it is eliminated from the system [49].
Through in vivo techniques, the bioavailability of a drug in a living system can be investigated. These methods consider the entire bioavailability process, including drug interactions, metabolism, elimination, and patient response. In vivo methods involve administering drugs to patients or animals and then analyzing samples of blood, urine, or other body fluids to determine the concentration of the drug over time [50].
With the advancements in science and technology, new methods and tools for evaluating drug bioavailability are being developed. Medical imaging techniques, such as computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography (PET), are some of the techniques that are currently being developed. These techniques enable the monitoring and tracking of drug distribution in the body, allowing for the observation of the drug’s path post-administration and the assessment of its concentration in various tissues and organs [51]. Other advanced techniques include pharmacogenetic studies, which deal with the impact of genetic differences on drug responses. They allow for the determination of the effect of genetic polymorphism in metabolic enzymes and transporters on drug bioavailability in different individuals [52]. Pharmacokinetic modeling, or computer simulation, has become a valuable medical tool that uses mathematical models and algorithms to predict drug performance in living organisms, considering factors like dosage, absorption, transport rates, and enzyme concentrations, thereby optimizing treatment efficacy [53].
4. Drugs with Poorly Described Bioavailability
Drugs with poorly described bioavailability are those for which there is limited information regarding the ADME process in the body. There may be various reasons for the lack of detailed data on drug bioavailability [16]. In this context, four distinct groups of drugs can be identified: (1) drugs with complex metabolism and elimination, (2) drugs with limited solubility, (3) drugs with specific absorption, and (4) other cases. Drugs with complex metabolism and elimination undergo intricate chemical metabolism and removal processes, influenced by factors such as interactions with other drugs, differences in gene-type metabolism, diseases, and the overall health of the patient [23].
Warfarin is a drug used for preventing and treating thrombosis. However, its metabolism is quite complex. It is mainly metabolized in the liver by cytochrome P450 enzymes through a series of steps involving hydroxylation, reduction, and conjugation. After metabolism, the drug is eliminated from the body as metabolites through the kidneys. However, the activity of cytochrome P450 enzymes varies widely due to genetic differences and interactions with other drugs and food.
Carbamazepine is a medication used to treat epilepsy, trigeminal neuralgia, and bipolar affective disorder. Certain enzymes, particularly CYP3A4 and CYP2C9 isoenzymes, metabolize the drug in the liver. Its metabolites are excreted in both urine and feces. Carbamazepine’s metabolism and bioavailability can be significantly affected by interactions with other drugs that inhibit or induce the cytochrome enzymes [54][55].
Digoxin is a medication used to treat heart failure and certain cardiac arrhythmias. Although it is primarily metabolized in the liver, it is eliminated from the body mainly through renal excretion. The metabolism of digoxin is complex, with glucuronidation being the primary metabolic pathway. However, the therapeutic window for digoxin is narrow, which means that even small changes in bioavailability and elimination can cause toxicity or a lack of effectiveness [56][57].
Drugs with low water solubility show difficulty dissolving in body fluids, which affects their bioavailability and therapeutic effectiveness due to the hydrophobicity of the drug or the formation of complexes with other substances [58][59]. Analgesics like diclofenac possess limited solubility in water, which adversely affects their absorption from the gastrointestinal tract and therefore, their bioavailability. Various strategies can be employed to increase the solubility and improve the absorption of these drugs. These may include modifying the formulation to obtain a more soluble form or using specific carriers that facilitate drug delivery to the site of action [60]. Certain antifungal medications, including itraconazole, exhibit a restricted capacity to dissolve in water, reducing their absorption in the gastrointestinal tract. To overcome this issue, solubility-enhancing substances are administered, or suitable formulations are developed to enhance the effectiveness and bioavailability of these drugs [61]. Paclitaxel is a common anticancer drug used to treat various malignancies in humans. However, it shows limited solubility in water, which makes it difficult to administer and absorb. To overcome this challenge, unique formulations enhance its solubility and delivery to the site of action [62].
Drugs with specific absorption are those whose absorption in the gastrointestinal tract depends on specific mechanisms or conditions. These drugs may be subject to interactions with other substances, pH changes, the presence of transporters, or specific absorption processes. One example of such a drug is levothyroxine, a synthetic hormone used to treat hypothyroidism. Its absorption depends on the presence of iodine in the gut. Iodine ions are essential for forming the active thyroid hormone (thyroxine—T4). Consequently, patients taking levothyroxine must take it on an empty stomach and avoid substances such as calcium, iron, or fiber that may affect the absorption of iodine and the drug itself [63].
Apart from to the examples outlined above, there are numerous other instances in which drug bioavailability is poorly defined or understood. Herbal treatments, anticancer medications, and novel medications are some examples of these. The bioavailability of certain plant-based drugs, such as herbal dietary supplements, can be inadequately described due to the complexity of their active ingredients, which can vary in their chemical form [64][65]. Hypericin is a natural compound in the St. John’s wort (Hypericum perforatum) plant. It is used for various health conditions, including treating depression and fighting against different viruses. However, the bioavailability of hypericin is not well understood because it is a complex chemical compound that can undergo several transformations in the body. Different forms of hypericin may include various pharmacokinetic properties, as well as bioavailability [64].
Data on bioavailability may be limited for new drugs that have not undergone comprehensive clinical trials. The absorption, distribution, metabolism, and elimination of a drug are all evaluated in basic pharmacokinetic studies. Nevertheless, data regarding bioavailability are scarce, particularly in large-scale clinical trials with a diverse patient group [16]. An example of a new drug with limited information regarding its bioavailability is tecovirimat (Tpoxx), an antiviral drug that has demonstrated efficacy in animal studies and has been approved by the Food and Drug Administration for the treatment of smallpox, a severe and life-threatening infection caused by the Variola virus of the Orthopoxvirus genus. It belongs to a group of drugs known as orthopoxvirus-specific antivirals. Tecovirimat is an investigational drug and is not currently approved for routine use. It is used in emergencies as part of preparatory measures against smallpox outbreaks [66].
5. Conclusions
Bioavailability data for many active compounds is sparse, despite substantial pharmacological study. These extensive pharmacokinetic studies are required for a broad list of drugs [67]. However, such studies are expensive and complicated; thus, few are performed, and therefore, few can be added to the bioavailability dataset [68]. Moreover, the pharmacokinetic characteristics of individuals vary greatly. Age, gender, genetics, health, and other parameters affect drug absorption and transport. However, a number of results obtained from such studies can be utilized to build generalized models showing the action of APIs in the human body [19].
It was established that drug bioavailability depends on administration [69]. Intravenous medications enter the bloodstream directly, while oral pharmaceuticals must pass through the digestive system and may be destroyed or absorbed incorrectly. Many bioavailability details remain undiscovered, despite broad studies for varied dosing techniques [70]. Since not all drug-food interactions are known, the need remains for further thorough studies of this aspect [71].
In regards to drug therapy safety, bioavailability studies determine doses to reduce dangerous blood active component concentrations, identify medication interaction risk factors, and improve safety [72]. Individualized treatment based on genetics, health, age, and drug interactions is possible because these studies disclose internal factors impacting drug absorption, distribution, and metabolism [73].
Medication bioavailability research is essential for treating rare and complex diseases. Understanding the active ingredient absorption derived from tablets, capsules, injections, and patches improves drug development. Understanding bioavailability improves drug use and health care by optimizing prescription design, treating rare disorders, and discovering new formulations [74].
References
- Herkenne, C.; Alberti, I.; Naik, A.; Kalia, Y.N.; Mathy, F.X.; Preat, V.; Guy, R.H. In vivo methods for the assessment of topical drug bioavailability. Pharm. Res. 2008, 25, 87–103.
- Olivares-Morales, A.; Hatley, O.J.; Turner, D.; Galetin, A.; Aarons, L.; Rostami-Hodjegan, A. The use of ROC analysis for the qualitative prediction of human oral bioavailability from animal data. Pharm. Res. 2014, 31, 720–730.
- Caldwell, J.; Gardner, I.; Swales, N. An introduction to drug disposition: The basic principles of absorption, distribution, metabolism, and excretion. Toxicol. Pathol. 1995, 23, 102–114.
- Martinez, M.N.; Amidon, G.L. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643.
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7.
- Patel, M.; Kumar, R.; Kishor, K.; Mlsna, T.; Pittman, C.U., Jr.; Mohan, D. Pharmaceuticals of emerging concern in aquatic systems: Chemistry, occurrence, effects, and removal methods. Chem. Rev. 2019, 119, 3510–3673.
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. Gut reaction: Impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug Discov. Today 2019, 24, 417–427.
- Wagner, J.G. History of pharmacokinetics. Pharmacol. Therapeut. 1981, 12, 537–562.
- Lin, L.; Wong, H. Predicting oral drug absorption: Mini review on physiologically-based pharmacokinetic models. Pharmaceutics 2017, 9, 41.
- Wang, Y.; Wang, Y.; Pi, C.; Feng, X.; Hou, Y.; Zhao, L.; Wei, Y. The influence of nanoparticle properties on oral bioavailability of drugs. Int. J. Nanomed. 2020, 15, 6295–6310.
- Alston, A.B.; Digigow, R.; Fluhmann, B.; Wacker, M.G. Putting square pegs in round holes: Why traditional pharmacokinetic principles cannot universally be applied to iron-carbohydrate complexes. Eur. J. Pharm. Biopharm. 2023, 188, 6–14.
- Hedaya, M.A. Basic Pharmacokinetics, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012.
- Wang, F.; Yang, G.; Zhou, Y.; Song, H.; Xiong, L.; Wang, L.; Shen, X. Pharmacokinetics of niazirin from Moringa oleifera Lam in rats by UPLC-MS/MS: Absolute bioavailability and dose proportionality. eFood 2022, 3, e39.
- Currie, G.M. Pharmacology, part 2: Introduction to pharmacokinetics. J. Nucl. Med. Technol. 2018, 46, 221–230.
- Tuntland, T.; Ethell, B.; Kosaka, T.; Blasco, F.; Zang, R.X.; Jain, M.; Hoffmaster, K. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 5, 174.
- Wei, M.; Zhang, X.; Pan, X.; Wang, B.; Ji, C.; Qi, Y.; Zhang, J.Z. HobPre: Accurate prediction of human oral bioavailability for small molecules. J. Cheminform. 2022, 14, 1–10.
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011.
- Kalaimathi, K.; Shine, K.; Gandhi, G.R.; Vijayakumar, S.; Ayyanar, M.; Amalraj, S.; Jose, J. Cyanobacterial metabolites as novel potential suppressors of breast cancer: A comparative in silico pharmacological assessment. Intell. Pharm. 2023, 1, 133–144.
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Müllertz, A. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280.
- Belayneh, A.; Molla, F. The effect of coffee on pharmacokinetic properties of drugs: A review. Biomed. Res. Int. 2020, 2020, 7909703.
- Sochacka, J.; Lipska, I. Rola α1-kwaśnej glikoproteiny surowicy krwi ludzkiej w procesie wiązania leków, sytuacja w Polsce i na świecie. Farmacja Polska 2014, 70, 55–62.
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308.
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Bi, H. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144.
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411.
- Settimo, L.; Bellman, K.; Knegtel, R.M. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014, 31, 1082–1095.
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokinetic modeling of drug absorption, distribution, metabolism, excretion, and toxicity. Expert Opin. Drug Met. 2021, 17, 1103–1124.
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Augustijns, P. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812.
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Yang, Z. Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381.
- Elliott, R.O.; He, M. Unlocking the Power of Exosomes for Crossing Biological Barriers in Drug Delivery. Pharmaceutics 2021, 13, 122.
- Kratzer, I.; Ek, J.; Stolp, H. The molecular anatomy and functions of the choroid plexus in healthy and diseased brain. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183430.
- Uchida, Y.; Goto, R.; Usui, T.; Tachikawa, M.; Terasaki, T. Blood-Arachnoid Barrier as a Dynamic Physiological and Pharmacological Interface between Cerebrospinal Fluid and Blood. In Drug Delivery to the Brain: Physiological Concepts, Methodologies and Approaches; Springer International Publishing: Cham, Germany, 2022; pp. 93–121.
- Kiecker, C. The origins of the circumventricular organs. J. Anat. 2018, 232, 540–553.
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliver. Rev. 2020, 165, 1–14.
- Villaseñor, R.; Lampe, J.; Schwaninger, M.; Collin, L. Intracellular transport and regulation of transcytosis across the blood–brain barrier. Cell. Mol. Life Sci. 2019, 76, 1081–1092.
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403.
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 1–24.
- Sánchez-Félix, M.; Burke, M.; Chen, H.H.; Patterson, C.; Mittal, S. Predicting bioavailability of monoclonal antibodies after subcutaneous administration: Open innovation challenge. Adv. Drug Deliver. Rev. 2020, 167, 66–77.
- Bannigan, P.; Aldeghi, M.; Bao, Z.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning directed drug formulation development. Adv. Drug Deliver. Rev. 2021, 175, 113806.
- Tanguay, M.; Girard, J.; Scarsi, C.; Mautone, G.; Larouche, R. Pharmacokinetics and comparative bioavailability of a levothyroxine sodium oral solution and soft capsule. Clin. Pharm. Dug Dev. 2019, 8, 521–528.
- Shariare, M.H.; Altamimi, M.A.; Marzan, A.L.; Tabassum, R.; Jahan, B.; Reza, H.M.; Kazi, M. In vitro dissolution and bioavailability study of furosemide nanosuspension prepared using design of experiment (DoE). Saudi Pharm. J. 2019, 27, 96–105.
- Alghamdi, M.A.; Fallica, A.N.; Virzì, N.; Kesharwani, P.; Pittalà, V.; Greish, K. The promise of nanotechnology in personalized medicine. J. Pers. Med. 2022, 12, 673.
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461.
- Australian Product Information Tivicay (dolutegravir) Film-Coated Tablets and TIVICAY PD (dolutegravir) Dispersible Tablets. Available online: https://www.tga.gov.au/sites/default/files/2022-08/auspar-tivicay-tivicay-pd-220705-pi.pdf (accessed on 10 October 2023).
- Shultz, M.D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs: Miniperspective. J. Med. Chem. 2018, 62, 1701–1714.
- U.S. FDA Approves GlaxoSmithKline’s HIV Drug Tivicay. Available online: https://www.reuters.com/article/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 (accessed on 5 October 2023).
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605.
- Ren, S.; Liu, M.; Hong, C.; Li, G.; Sun, J.; Wang, J.; Xie, Y. The effects of pH, surfactant, ion concentration, coformer, and molecular arrangement on the solubility behavior of myricetin cocrystals. Acta Pharm. Sin. B 2019, 9, 59–73.
- Santbergen, M.J.; Van der Zande, M.; Gerssen, A.; Bouwmeester, H.; Nielen, M.W. Dynamic in vitro intestinal barrier model coupled to chip-based liquid chromatography mass spectrometry for oral bioavailability studies. Anal. Bioanal. Chem. 2020, 412, 1111–1122.
- Attwa, M.W.; AlRabiah, H.; Mostafa, G.A.; Kadi, A.A. Development of an LC-MS/MS method for quantification of sapitinib in human liver microsomes: In silico and in vitro metabolic stability evaluation. Molecules 2023, 28, 2322.
- Shinha, K.; Nihei, W.; Ono, T.; Nakazato, R.; Kimura, H. A pharmacokinetic–pharmacodynamic model based on multi-organ-on-a-chip for drug–drug interaction studies. Biomicrofluidics 2020, 14, 044108.
- Perez-Medina, C.; Teunissen, A.J.; Kluza, E.; Mulder, W.J.; Van der Meel, R. Nuclear imaging approaches facilitating nanomedicine translation. Adv. Drug Deliver. Rev. 2020, 154, 123–141.
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643.
- Dałek, P.; Drabik, D.; Wołczańska, H.; Foryś, A.; Jagas, M.; Jędruchniewicz, N.; Langner, M. Bioavailability by design—Vitamin D3 liposomal delivery vehicles. Nanomed. Nanotechnol. Biol. Med. 2022, 43, 102552.
- Li, H.; Zhang, M.; Xiong, L.; Feng, W.; Williams, R.O., III. Bioavailability improvement of carbamazepine via oral administration of modified-release amorphous solid dispersions in rats. Pharmaceutics 2020, 12, 11–1023.
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug–drug interaction potential: A physiologically based pharmacokinetic modeling approach. Pharmaceutics 2021, 13, 270.
- Hsin, C.H.; Stoffel, M.S.; Gazzaz, M.; Schaeffeler, E.; Schwab, M.; Fuhr, U.; Taubert, M. Combinations of common SNPs of the transporter gene ABCB1 influence apparent bioavailability, but not renal elimination of oral digoxin. Sci. Rep. 2020, 10, 12457.
- Ibrahim, N.A.M. An up-to-date review of digoxin toxicity and its management. Int. J. Res. Pharm. Pharm. Sci. 2019, 4, 59–64.
- Pawar, S.R.; Barhate, S.D. Solubility enhancement (Solid Dispersions) novel boon to increase bioavailability. J. Drug Deliv. Ther. 2019, 9, 583–590.
- Kareem, S.H.K.A. Quality by Design Approach for Bioavailability Enhancement of Some Hydrophobic Drugs. Available online: https://shodhgangotri.inflibnet.ac.in/bitstream/20.500.14146/13393/1/final%20synopsis%20corrected.pdf (accessed on 5 October 2023).
- Pireddu, R.; Schlich, M.; Marceddu, S.; Valenti, D.; Pini, E.; Fadda, A.M.; Sinico, C. Nanosuspensions and microneedles roller as a combined approach to enhance diclofenac topical bioavailability. Pharmaceutics 2020, 12, 1140.
- Sardana, K.; Mathachan, S.R. Super bioavailable itraconazole and its place and relevance in recalcitrant dermatophytosis: Revisiting skin levels of itraconazole and minimum inhibitory concentration data. Indian Dermatol. Online J. 2021, 12, 1.
- Bardelmeijer, H.A.; Beijnen, J.H.; Brouwer, K.R.; Rosing, H.; Nooijen, W.J.; Schellens, J.H.; van Tellingen, O. Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin. Cancer Res. 2000, 6, 4416–4421.
- Virili, C.; Brusca, N.; Capriello, S.; Centanni, M. Levothyroxine therapy in gastric malabsorptive disorders. Front. Endocrinol. 2021, 11, 621616.
- Oglah, M.K.; Bashir, M.K.; Mustafa, Y.F. Hypericin and its analogues: A review of their biological activities. Turk. J. Field Crops 2021, 26, 259–269.
- Lin, Y.; Li, Y.; Zeng, Y.; Tian, B.; Qu, X.; Yuan, Q.; Song, Y. Pharmacology, toxicity, bioavailability, and formulation of magnolol: An update. Front. Pharmacol. 2021, 12, 632767.
- O’Laughlin, K.; Tobolowsky, F.A.; Elmor, R.; Overton, R.; O’Connor, S.M.; Damon, I.K.; Veillard, M. Clinical use of tecovirimat (Tpoxx) for treatment of monkeypox under an investigational new drug protocol—United States, May–August 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1190.
- Bohn, T.; Desmarchelier, C.; Dragsted, L.O.; Nielsen, C.S.; Stahl, W.; Rühl, R.; Borel, P. Host-related factors explaining interindividual variability of carotenoid bioavailability and tissue concentrations in humans. Mol. Nutr. Food Res. 2017, 61, 1600685.
- Deore, A.B.; Dhumane, J.R.; Wagh, R.; Sonawane, R. The stages of drug discovery and development process. Asian J. Pharm. Res. Dev. 2019, 7, 62–67.
- Trucillo, P. Drug carriers: Classification, administration, release profiles, and industrial approach. Processes 2021, 9, 470.
- Landersdorfer, C.B.; Gwee, A.; Nation, R.L. Clinical pharmacological considerations in an early intravenous to oral antibiotic switch: Are barriers real or simply perceived? Clin. Microbiol. Infec. 2023, 29, 1120–1125.
- Koziolek, M.; Alcaro, S.; Augustijns, P.; Basit, A.W.; Grimm, M.; Hens, B.; Corsetti, M. The mechanisms of pharmacokinetic food-drug interactions–A perspective from the UNGAP group. Eur. J. Pharm. Sci. 2019, 134, 31–59.
- Drenth-van Maanen, A.C.; Wilting, I.; Jansen, P.A. Prescribing medicines to older people—How to consider the impact of ageing on human organ and body functions. Brit. J. Clin. Pharmacol. 2020, 86, 1921–1930.
- Baillie, T.A.; Cayen, M.N.; Fouda, H.; Gerson, R.J.; Green, J.D.; Grossman, S.J.; Shipley, L.A. Drug metabolites in safety testing. Toxicol. Appl. Pharm. 2002, 182, 188–196.
- May, M.; Schindler, C.; Engeli, S. Modern pharmacological treatment of obese patients. Ther. Adv. Endocrinol. Metab. 2020, 11, 1–19.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Revisions:
2 times
(View History)
Update Date:
03 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No