Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alicja Nowaczyk and Version 2 by Lindsay Dong.
Drug bioavailability is a crucial aspect of pharmacology, affecting the effectiveness of drug therapy. Understanding how drugs are absorbed, distributed, metabolized, and eliminated in patients’ bodies is essential to ensure proper and safe treatment. In addition to biochemical activity, bioavailability also plays a critical role in achieving the desired therapeutic effects. This may seem obvious, but it is worth noting that a drug can only produce the expected effect if the proper level of concentration can be achieved at the desired point in a patient’s body. Given the differences between patients, drug dosages, and administration forms, understanding and controlling bioavailability has become a priority in pharmacology.
- bioavailability
- pharmacokinetics
- therapeutic effectiveness
- pharmaceutical innovations
1. Introduction
Drug bioavailability plays a crucial role in the effectiveness of pharmacological therapy. It determines the degree and rate at which drug-active substances are absorbed into the bloodstream after oral, topical, parenteral, and rectal administration [1]. In practice, bioavailability indicates the amount of the administered dose of a drug reaching the bloodstream in the form of the active ingredient which is then available to the body to produce a therapeutic effect [2].
Bioavailability is affected by variety of factors, including the medication’s physicochemical properties, the mode of administration, interactions with other substances, absorption, hepatic metabolism, and excretion [3][4][3,4]. The bioavailability of the active pharmaceutical ingredients (API) corresponds to the dose entering the bloodstream and consequently, its effectiveness [2]. For this reason, a drug’s bioavailability must be considered when designing a therapy and dosage. The efficient adjustment of an administered dosage requires knowledge of the API’s absorption, transport mechanism, metabolism, and elimination from the system [5]. Conversely, the ineffective administration of medications adds to the escalation of superfluous drug use. From this perspective, the significance of bioavailability becomes even more significant when it comes to patient safety and treatment efficacy [6]. Improving medication bioavailability can be accomplished by using suitable drug delivery methods, modifying drug formulations, optimizing dosages, identifying and controlling factors that decrease bioavailability, and monitoring blood drug levels resulting from dose adjustments. The monitoring of bioavailability can provide insights into drug interactions, as well as enable the development of tailored treatment plans for patients with liver or intestinal dysfunction [7]. Safe drug therapy requires good bioavailability. Insufficient bioavailability can reduce therapy efficacy, whereas excessive medication concentrations can produce toxicity and side effects [8]. Nevertheless, drug bioavailability is only one of many factors impacting the efficacy of drug therapy [9]. Currently, the field of pharmaceutical research and development is facing various challenges, one of which is the optimization of medication bioavailability. This optimization is pursued with the goal of enhancing the safety and efficacy of treatments [10].
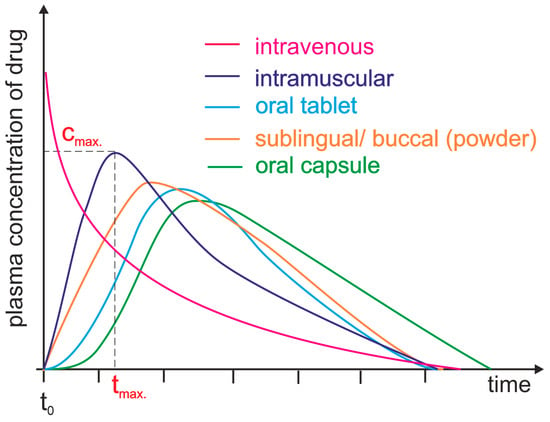
2. Bioavailability of Drugs: Basic Concepts and Controlling Factors
The bioavailability of a drug describes the level of absorption of the active substance contained therein and the speed at which it is absorbed from the form administered to the patient, becoming available in the targeted location of the body, usually in the bloodstream. The bioavailability of a drug determines the proportion of the active substance available in the body in relation to its amount in the drug [11][15]. Drug bioavailability measurements allow for the assessment of the absorption efficiency of a drug, including its absolute and relative bioavailability, the time to reach maximum concentration, and the area under the concentration–time curve [12][16]. Absolute bioavailability is defined as a measurement that determines the percentage of the active substance entering the bloodstream after administration of a drug when the reference standard is an intravenous dose. It expresses the efficiency of the absorption of the active substance by the body. It is usually less than 100%, since not all of the active substance is absorbed in the gastrointestinal tract [13][17]. Relative bioavailability is expressed as the ratio of the bioavailability of two dosage forms of the same drug (Figure 1). The time to reach maximum concentration (tmax) measures the time it takes for an active ingredient to reach its highest concentration in the blood after drug administration. tmax is an important parameter because it can affect the rate of action of a drug. The area under the curve (AUC) measures the total amount of an active substance absorbed and available in the bloodstream as a function of time. The AUC is used to assess the total exposure of the body to an active substance (Figure 1) [14][19]. Measurements of drug bioavailability are essential for evaluating the pharmacokinetics and pharmacodynamics of drugs and for determining appropriate therapeutic doses. They are also crucial for developing new drugs and evaluating the efficacy of different administration forms [12][15][16,20].Figure 1. Plasma level time curves for different types of drug administration. The drug is delivered directly into the systemic circulation via intravenous injection, ensuring 100% bioavailability and immediate achievement of maximum plasma concentration (cmax, tmax = 0 min). For other parenteral routes, such as subcutaneous and intramuscular injections, most drugs show between 60 and 100% bioavailability due to little or no metabolism in the skin or muscle. However, the time to reach maximum plasma concentration (tmax) is significantly longer than that achieved by intravenous administration. Orally administered drugs achieve a bioavailability level substantially lower than 100% due to incomplete absorption and/or elimination during the first pass through the liver. Additionally, due to the indirect path to the plasma, they are characterized by a long time lag. Different dosage forms may result in differences in cmax and tmax.
The ADME processes affect drug absorption, distribution, metabolism, and elimination in the body [16][17][21,22].
Absorption is the passing of a drug from the administration site (e.g., gastrointestinal tract, skin, respiratory system) into the bloodstream [18][23]. When administered orally, the drug must be absorbed from the gastrointestinal tract into the bloodstream to achieve its effect. Various factors can determine absorption, such as dosage form, the presence of food, the environment’s pH, or interactions with other substances [19][24].
Distribution is defined as the spread of an active compound throughout the body, leading to the presence of the drug in various tissues and organs after its absorption into the bloodstream. Distribution depends on several factors, such as vascular resistance, distribution volume, degree of drug binding to plasma proteins, and penetration of tissue barriers (e.g., permeability of the blood–brain barrier) [20][25]. Once a substance enters the body, part of it can bind to proteins in the blood, mainly albumin (acidic and neutral molecules, such as vitamins, drugs, and their metabolites), but also other proteins, such as acidic α1-glycoprotein and lipoproteins, as well as gamma globulins. Drug–protein complexes are too large to passively penetrate cell membranes, which affects drug distribution in the body. For the desired pharmacological effect to be achieved, most of the drug must be in free (unbound) form, since they can only interact with receptors in this form. If two or more drugs bind to the same plasma protein, they compete for a binding site, affecting their biological availability, as well as their effects [21][26].
The process by which the body, with the assistance of enzymes, transforms a drug into different substances—known as metabolites—is referred to as drug metabolism [22][33]. The liver is the most critical site of drug metabolism. This process aims to convert the drug into more soluble substances, which are more efficiently eliminated from the body, and to manage its concentration and activity [23][34].
Eliminating drugs from the body involves the removal of both the drug and its metabolites from the organism. The two main routes of elimination are excretion through urine and secretion via bile. The pharmacokinetic processes play a vital role in determining the appropriate drug doses, determining the optimal timing of administration, and assessing the therapeutic effectiveness. [14][19].
Factors influencing the bioavailability of drugs can be divided into four main categories: physicochemical agents, biological agents, pharmaceutical agents, and patient factors. The physicochemical properties of a drug are important for its bioavailability. Examples of physicochemical factors include drug solubility in the digestive environment, chemical stability, lipophilicity, ionizability, and pharmaceutical form.
A drug must be soluble in the intestinal environment to be absorbed across biological membranes. Additionally, the ability of a drug to penetrate the membrane can be altered by its ionic form, which depends on the environment’s pH [24][35]. Drug molecules can ionize over different pH ranges, and the acid–base dissociation constant (pKa) is adopted as an universal measure of ionization. The pH range ϵ (2–12) indicates the point at which 63% of drugs ionize, according to the World Narcotics Index. About 43% and 12% of drugs, respectively, contain a single primary or acid center. The ionization process can significantly impact the properties of drug absorption, distribution, metabolism, excretion, and toxicity in vivo. Changing a drug’s ionic form (Figure 2) can affect its action, absorption, and therapeutic efficacy, depending on the environment’s pH [25][26][36,37].
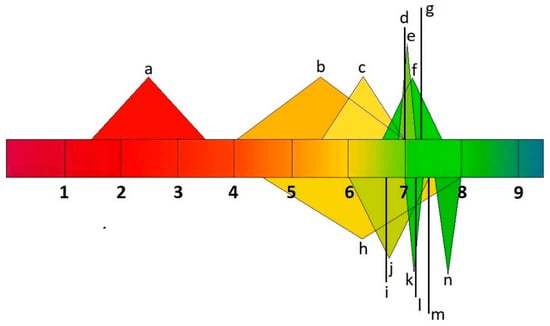
Figure 2. pH values of human body fluids a: stomach; b: small intestine; c: large intestine; d: liver; e: muscle; f: uterus; g: testis; h: bladder; i: lungs; j: saliva; k: kidneys; l: brain, heart, and spleen; m: bone; n: pancreas.
The bioavailability of drugs can be significantly affected by various biological processes occurring in the body, such as gastric acidity, organ blood flow, digestive enzyme activity, intestinal microflora, and biological barriers. Gastric acidity, for example, may influence drug distribution and solubility. Furthermore, the activity of digestive enzymes in the gut can either reformulate the drug or reduce its availability, influencing its effectiveness [27][38]. Biological barriers, such as the intestinal cell membrane and blood–brain barrier, can limit the penetration of a drug into the bloodstream [28][39]. Biological barriers protecting the system against pathogens complicate drug delivery and distribution [29][40]. The adult brain possesses five barrier interfaces that regulate molecular movement into the brain parenchyma. These are the blood–brain barrier (BBB) the blood–cerebrospinal fluid barrier (BCSFB) [30][41], the blood–arachnoid barrier (BAB) [31][42], the circumventricular organs (CVOs) [32][43], and the ependyma [33][44]. The blood–brain barrier (BBB) is created by a tight structure of endothelial cells (ECs) joined together by protein couplings. These cells line the cerebral microvessels, separating the blood from the brain’s interstitial fluid [34][45]. The choroid plexus epithelium is between the blood and the ventricular cerebrospinal fluid (CSF) and forms the blood–CSF barrier. The epithelium between the blood and the subarachnoid CSF forms the arachnoid barriers. These three barrier layers limit and regulate molecular exchange at the interface between the blood and neural tissue or its fluid spaces. [35][46]. The inherent function of biological barriers impedes drug delivery and uptake, preventing effective therapeutic interventions. Biological barriers hinder treatment options and reduce the bioavailability of drugs in areas protected by barriers, which can ultimately lead to increased drug resistance [36][47].
Pharmaceutical factors encompass the technologies used to create a drug. These include drug formulation, excipients, formulation methods, and drug release techniques [37][48]. The main objective of any drug delivery system (DDS) is to maintain the desired therapeutic effect by providing and sustaining adequate drug concentration at the target site in the body. This involves enhancing drug efficacy; resolving issues related to solubility, low bioavailability, and poor in-body distribution; and minimizing side effects.
The drug formulation process often involves combining inactive ingredients and additional substances with APIs to produce drug products with specific characteristics. Improving this process to achieve an optimal drug formulation can involve various objectives such as increasing efficacy, extending the duration of therapeutic effects, reducing adverse effects, prolonging the shelf life of active ingredients, and enhancing compatibility with patient intake patterns [1]. APIs can be formulated using different material combinations, including neutral boosters such as polymers, lipids, surfactants, and other active ingredients, depending on the desired delivery method and specific application requirements. Such formulations are made possible by utilizing various types of delivery systems, including different kinds of microparticles (MPs), nanoparticles (NPs), and complex multi-component systems [2][3][4][5][2,3,4,5]. Typical practices involving these delivery mechanisms are often evolving, resulting in the development of drug products in various forms, such as solids, liquids, or non-oral administration methods [38][50].
Drug forms such as tablets, capsules, granules, powders, suspensions, solutions, emulsions, inserts, ointments, inserts, aerosols, patches, and transdermal systems affect the drug’s dissolution rate and absorption [39][51]. Excipients, such as binders, solvents, and stabilizers, can affect a drug’s bioavailability through interactions with the drug or changes in its solubility [40][52]. Drug release techniques can control the active ingredient’s release rate, which affects its availability and action [37][48]. Acetylsalicylic acid, commonly known as aspirin, is available in various administration forms. It can be taken orally as tablets in enteral, enteric-coated, effervescent, and controlled-release forms.
Patient factors refer to individual patient characteristics that can affect the bioavailability of a drug [41][55]. These include age, gender, genotype, health status, and diet. The drug absorption, metabolism, and elimination processes may differ depending on age. Children and the elderly may exhibit differences in metabolic enzymes, renal function, and blood flow, which can affect the bioavailability of drugs.
The protein known as P-glycoprotein (P-gp) plays a critical role in creating barriers within cells, particularly in the endothelial cells of the blood vessels. Its primary role is to prevent the entry of various substances, including drugs, into neural tissue by removing them from endothelial cells and returning them into the bloodstream. P-gp is a multidrug transporter that can recognize many compounds with different chemical structures and molecular weights (ranging from 330 to 4000 Da) [42][61]. It can transport hydrophobic and inert substances, as well as cations, but it cannot transport anions. The log p value of approximately 2.2 for DTG [43][62] indicates that it is only partially subject to bioaccumulation due to its moderate hydrophobicity [44][63]. P-gp is a protein crucial in transporting substances into and out of cells. Dolutegravir (DTG) is a substance for which P-gp is particularly important. Studies have shown that when DTG enters endothelial cells from the blood, it is pumped back into the bloodstream due to P-gp activity. However, disruption of the blood–brain barrier caused by HIV can lead to the dysfunction of P-gp, making it easier for drugs like DTG to penetrate brain tissues. This can result in higher concentrations of DTG in the brain, leading to unwanted side effects such as insomnia and headaches [45][64]. It is important to note that P-gp is present in tissues with a secretory function, such as the small intestine, liver, and kidney. If there is a pathological dysfunction of the P-gp protein, it can result in increased symptoms of dysfunction in these tissues. Recent studies reveal that P-gp triggers the production of effector T cells after viral infection.