Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Broggini Gianluigi | -- | 11431 | 2023-11-17 16:27:04 | | | |
| 2 | Jason Zhu | Meta information modification | 11431 | 2023-11-20 06:15:14 | | | | |
| 3 | Jason Zhu | Meta information modification | 11431 | 2023-12-29 07:05:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Papis, M.; Foschi, F.; Gianluigi, B.; Loro, C.; Colombo, S. Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. Encyclopedia. Available online: https://encyclopedia.pub/entry/51759 (accessed on 23 July 2026).
Papis M, Foschi F, Gianluigi B, Loro C, Colombo S. Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. Encyclopedia. Available at: https://encyclopedia.pub/entry/51759. Accessed July 23, 2026.
Papis, Marta, Francesca Foschi, Broggini Gianluigi, Camilla Loro, Sara Colombo. "Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds" Encyclopedia, https://encyclopedia.pub/entry/51759 (accessed July 23, 2026).
Papis, M., Foschi, F., Gianluigi, B., Loro, C., & Colombo, S. (2023, November 17). Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. In Encyclopedia. https://encyclopedia.pub/entry/51759
Papis, Marta, et al. "Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds." Encyclopedia. Web. 17 November, 2023.
Copy Citation
The functionalization of unactivated substrates through the combination of copper catalysts and hypervalent iodine reagents represents a versatile tool in organic synthesis to access various classes of compounds. The hypervalent iodine derivatives can be used simply as oxidizing agents to regenerate the catalytic species or they can associate the functionalization of the starting material.
copper catalysis
hypervalent iodine
difunctionalization reactions
1. Introduction
Copper-catalyzed oxidative coupling reactions involving multiple unactivated C-C bonds have emerged as an important strategy for rapidly increasing molecular complexity in organic synthesis. Compared to the classic coupling reactions, this approach avoids the need for pre-functionalized reactants, even if the success of the course depends on the ability of an appropriate oxidizing agent to restart the catalytic cycle. In this context, the hypervalent iodine reagents have gained a prominent role due to their unique features as non-toxic, relatively cheap, easy to handle, readily available, and with low environmental impact compared to the toxic heavy metal-based oxidants and expensive organometallic catalysts.
Today, hypervalent iodine compounds occupy a leading role in organic synthesis since they can be used as efficient reagents or simply as oxidizing agents [1][2][3][4][5][6][7][8][9][10][11][12]. Organic derivatives of iodine in the (III) and (V) oxidation states are usually used in organic synthesis as reagents for various selective oxidative transformations of complex organic molecules.
Hypervalent iodine reagents make the approach to organic synthesis more flexible as they allow the reversal of the normal polarity of functional groups, leading to their umpolung. They behave as electrophilic synthons, accessible from nucleophiles by exploiting the electrophilicity of the iodine atom and the reactivity of the hypervalent bond. Since the publication of the pioneering works in this field, both iodine and iodonium salts have assumed a privileged position for the possibility of transforming many nucleophiles into electrophilic synthons. However, although highly reactive, they can be unstable in the presence of strong bases, transition metals or heating, becoming unsuitable for many applications. To overcome this drawback, hypervalent cyclic iodine reagents are used, as the inclusion of the iodine atom in a heterocycle has shown greater stability than the acyclic equivalents [13][14]. As a mild oxidizing agent, Dess-Martin periodinane is used for the conversion of alcohols to aldehydes and ketones [15][16][17][18], while 2-iodoxybenzoic acid allows the oxidation of the benzylic position or the introduction of α,β-double bonds in carbonyl compounds [19][20]. The excellent reactivity of iodine(III) compounds due to their electrophilic character has been exploited to carry out alkynylation, azidation, cyanation and trifluoromethylation processes, also on the basis of the further opportunity to modulate the reactivity through the trans-effect in the hypervalent bond [21]. The availability of ethinyl [22][23], azido [24] and cyano-benziodoxolones [25] (EBX, EBX, and CBX, respectively) has allowed reactions of multifunctionalization to develop with the formation of more than one bond in a single transformation. Particularly interesting are the intramolecular oxidative transition metal-catalyzed processes of C-H functionalization that allow to associate the cyclization by alkoxylation and amination of multiple carbon–carbon bonds with the introduction of functional groups which provide direct access to functionalized heterocyclic systems. Conversely, diaryliodonium reagents have been recognized as efficient alternatives to transition metals. The most important and widespread application in the field of organic synthesis concerns arylation reactions using diaryliodonium salts. Due to the highly electron-deficient nature of diaryliodonium salts at the iodine core and the excellent behaviour as the leaving group of iodobenzene, they serve as versatile arylating agents with a variety of nucleophiles. Diaryliodonium salts with a halide anion are poorly soluble in most of the organic solvents, while triflates and tetrafluoroborates have better solubility.
2. Hypervalent Iodine as Reagent in C-C Bond Formation
2.1. Arylation Reactions
Biaryl and aryl-heteroaryl moieties are ubiquitous substructures in several compounds of primary interest in organic chemistry. Thus, arylation chemistry always demands new direct and selective procedures, possibly involving C-H bonds [26][27][28].
The combination of a copper catalyst and aryl hypervalent iodine derivatives was proven to be an innovative tool for the unusual meta-selective C–H bond arylation of electron-donor substituents in aromatic rings. The presence of a suitable carbonyl group as a directing group is essential to achieve meta-selectivity. In 2009, Gaunt’s group described the meta-arylation of pivanilides combining the use of catalytic Cu(OTf)2 with Ph2IOTf, Ph2IBF4 or [Mes-I-Ar]OTf (Scheme 1) [29]. The proposed mechanism, also studied theoretically by DFT calculations [30], involves the high electrophilic aryl-Cu(III) specie I as the key intermediate.
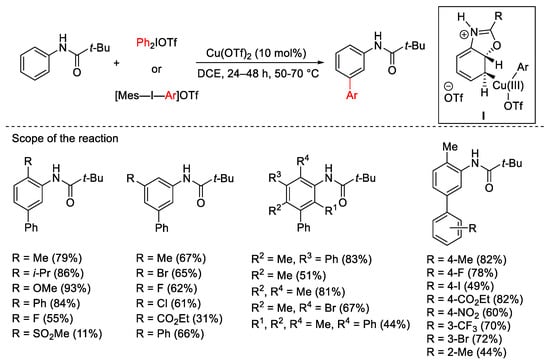
Scheme 1. Cu(OTf)2-catalyzed arylation of pivanilides.
Two years later, the selective arylation of the meta aromatic C–H sites was extended to the α-aryl carbonyl compounds (Scheme 2) [31]. A range of diaryliodonium salts was used with variously substituted arenes providing selective meta-arylation of the position bearing the methylene-carbonyl moiety. Among the carbonyl compounds, Weinreb amides showed better results and the enantiopure substrates reacted with total retention of the configuration.
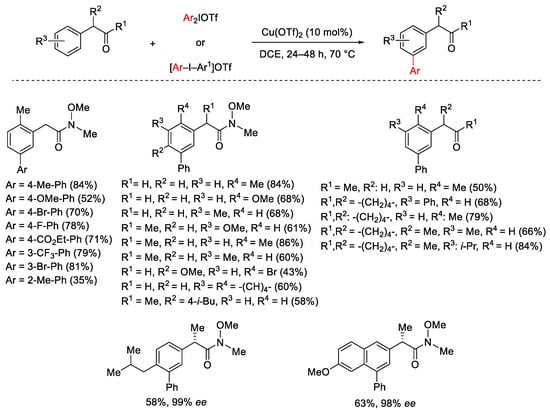
Scheme 2. Cu(OTf)2-catalyzed arylation of α-aryl-substituted carbonyl compounds.
In 2011, Gaunt and co-workers reported the para-selective arylation of electron-rich arenes, such as aniline and phenol derivatives, with a range of diaryliodonium salts in the presence of Cu(OTf)2 as the catalyst (Scheme 3) [32]. The reaction follows a Friedel–Craft-type mechanism and, whereas phenol derivatives are arylated without the need of additives, the arylation of anilines requires the use of 2,6-di-tert-butylpyridine (dtbpy) as a base to neutralize the TfOH generated in situ. The reaction also takes place on para-substituted arenes with ortho-orientation.
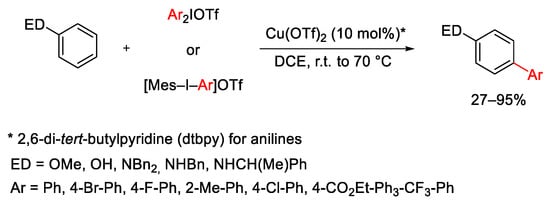
Scheme 3. Cu(OTf)2-catalyzed arylation of electron-rich arenes.
Diaryliodonium salts can be successfully used in copper-catalyzed C-H arylation of heteroarenes. In 2008, Gaunt and co-workers proposed the selective C3-arylation of indoles in mild conditions (Scheme 4) [33]. The reaction occurs on N-methyl and free (NH)-indoles, and the use of 2,6-di-tert-butylpyridine (dtbpy) as an additive avoids the competitive acid-catalyzed dimerization of the indole nucleus.
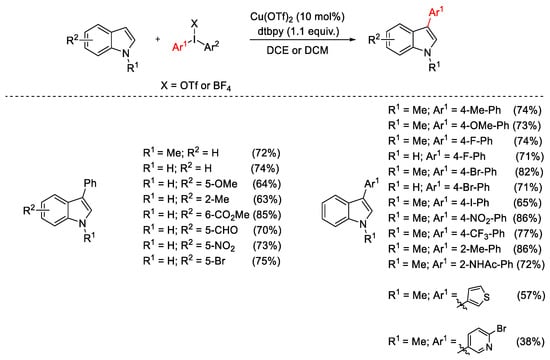
Scheme 4. Cu(OTf)2-catalyzed C3-arylation of indoles.
Conversely, operating in the same conditions, a selective C2-arylation was observed starting from N-acetylindoles (Scheme 5).
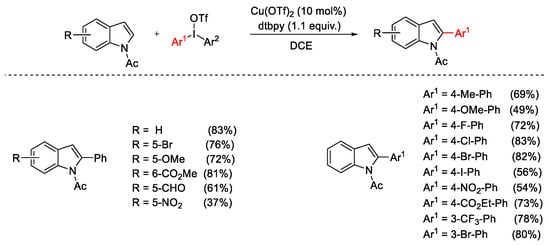
Scheme 5. Selective Cu(OTf)2-catalyzed C2-arylation of N-acetylindoles.
The proposed mechanism begins with the oxidative addition of the diaryliodonium salt to the Cu(I)-OTf species generated in situ, giving an electrophilic aryl-Cu(III) intermediate with concomitant loss of aryl iodide (Scheme 6). The more nucleophilic C3-indolyl position furnishes the Cu(III)-complex II, which evolves by base-promoted aromatization delivering the intermediate III. Finally, reductive elimination provides the C-C bond with regeneration of the Cu(I)-catalyst. The presence of the N-acetyl group plausibly renders the imino intermediate IV more suitable to accept the migrating C-Cu bond by coordination of the Cu(III) centre to the carbonyl group.
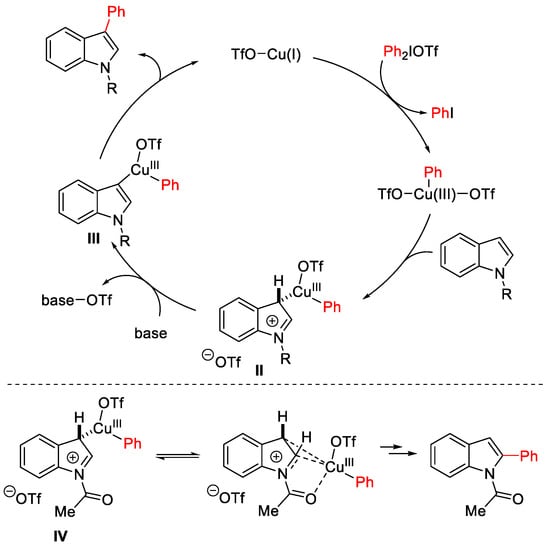
Scheme 6. Proposed mechanism for the selective Cu(OTf)2-catalyzed C3 arylation of indoles vs. C2-arylation of N-acetylindoles.
The use of a Cu-(bis)oxazoline complex as catalyst and the presence of a benzyl group into the hypervalent iodine reagents, allowed efficient enantioselective C-H arylation of 2-arylindoles, providing the atroposelective synthesis of the corresponding indoles 3 with excellent stereoselectivity (Scheme 7). DFT calculations and wave function analysis showed the key role of the conformation of the intermediate V. Due to the steric hindrance of the chiral ligand, the indole substrate 1 prefers to react from the right side forming a “sandwich”-like structure in which 1 was between the benzene ring of the ligand and the benzene ring of the benzyl group of the reagent 2 [34].
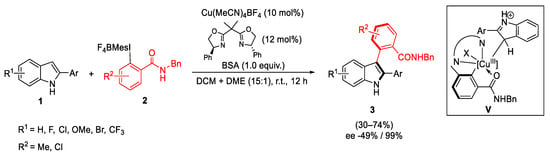
Scheme 7. Atroposelective synthesis of 2,3-diarylindoles.
Arene and heteroarene arylation found application in the total synthesis of dictyodendrin B, which was performed by sequential procedures involving, among others, C3 and C7 C-H arylation of the indole nucleus [35].
The selective C5 and C6 C-H arylation of indoles, proposed by Shi and co-workers, can be achieved depending on the presence of sterically hindered substituents on the substrates. The C5 arylation occurs on 3-pivaloyl-substituted indoles when treated with copper(I) thiophene-2-carboxylate (CuTC) as the catalyst, and 4,4′-di-tbutyl-2,2′-bipyridine (dtpby) and Ph2IOTf or ArMesIOTf as the arene source (Scheme 8) [36]. Conversely, the C6 C-H arylation is possible when the removable P(O)tBu2 directing group is present at N-position [37]. The reaction between these substrates and different diaryliodonium triflates in the presence of catalytic CuO proceeds in mild conditions. The initial coordination of the aryl-Cu(III) intermediate to the phosphoryl oxygen of the directing group generates the less hindered complex VI from which the transfer of the aryl group at C6 position arises from a Heck-type process (intermediate VII). A final base-assisted β-elimination furnishes the arylated products and regenerates the Cu(I)-catalyst.
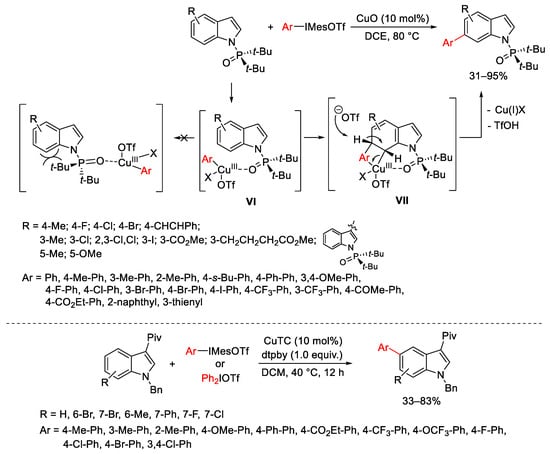
Scheme 8. Selective C5- and C6-arylation of substituted indoles.
In general, the use of diaryliodonium salts generates one equivalent of an iodoarene as a side-product, a significant problem in the context of atom economy. The possibility to use the aryliodonium salts for a dual arylation reactions in one step with no waste of arene residues was a stimulating challenge. This approach was investigated on indoles in a Cu-catalyzed tandem arylation reaction (Scheme 9). The tandem reaction, which occurs through an initial C-H arylation followed by N-arylation, was applied using symmetrical diaryliodonium salts and extended to unsymmetrical analogues. In this case, due to the very sensitive N-arylation to the steric hindrance, the hindered aryl iodides were not productive. Better results were obtained with phenyl-uracil iodonium triflate, producing 3-aryl-N-uracil indoles 4 [38].
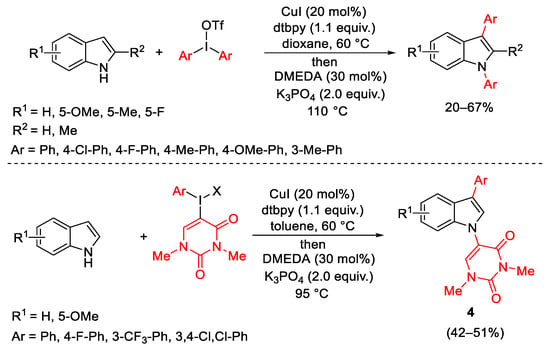
Scheme 9. Tandem diarylation of indoles.
Regarding the stereoselective arylation of enolizable aldehydes, MacMillan used diaryliodonium salts in combination with copper and imidazolyl organic catalysts, obtaining enantioselective α-aryl aldehydes performing the reaction in mild reaction conditions (Scheme 10) [39].
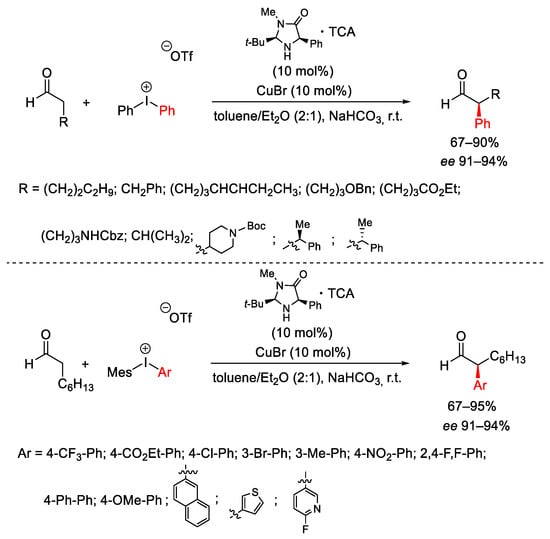
Scheme 10. Stereoselective arylation of enolizable aldehydes.
The reaction began with formation of chiral enamine 6 arising from the aldehyde substrate and the chiral amine 5 and run through the electrophilic Ar-Cu(III)-OTf-Br system VIII obtained by oxidative addition of Cu(I) into the C-I bond of the diaryliodonium triflate system (Scheme 11). This latter, highly electrophilic, coordinates to the less hindered face of the enamine to generate the π-copper complex IX, which in turn evolves to the intermediate X. The following reductive elimination affords the optically active iminium salt XI regenerating the Cu(I)-catalyst. The hydrolysis of XI furnishes the α-aryl aldehydes as final products beside the organocatalyst 5. The protocol was applied also to the synthesis of (S)-ketoprofen, obtained in 71% yield and 92% ee.
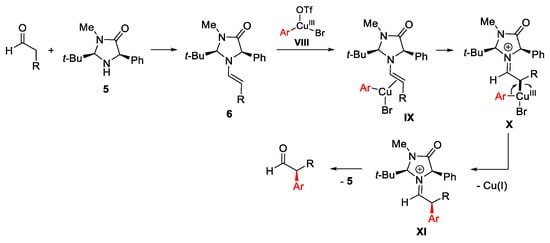
Scheme 11. Proposed mechanism for stereoselective arylation of enolizable aldehydes.
An efficient construction of α-aryl-β-substituted cyclic ketones was realized with excellent diastereoselectivity, as a one-pot procedure consisting in Michael addition/α-arylation reactions (Scheme 12) [40]. Gilman reagents as the nucleophilic Michael donor and electrophilic diaryliodonium salts as the trapping reagent of the substituted enolate intermediate were proven as suitable reagents. The proposed reaction mechanism suggested the formation of an intermediate Cu(III)-Ar, able to react with a Michael addition enolate intermediate and subsequent phenyl group transfer, forming the product and regenerating the Cu(I) catalyst.
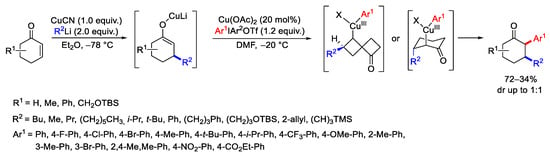
Scheme 12. Synthesis of α-aryl-β-substituted cyclic ketones.
In 2011, Gaunt developed a system for an efficient enantioselective α-arylation of N-acyloxazolidinones using chiral copper(II)-bisoxazoline complexes and diaryliodonium salts under mild conditions (Scheme 13). In this synthetic strategy, the silylketeneimide 7 derived from N-acyloxazolidinones was used as an enolate equivalent in the reaction with the preformed chiral catalyst XII, generated from Cu(OTf)2 and (R,R)-diphenyl-bisoxazoline [41].
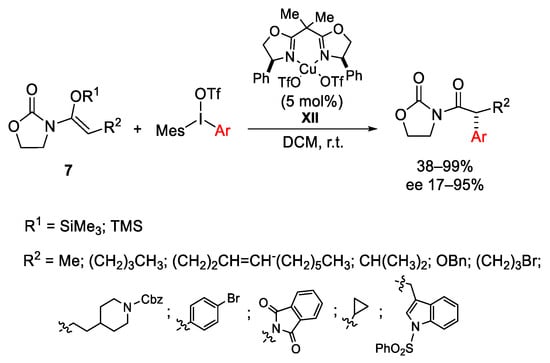
Scheme 13. Enantioselective α-arylation of N-acyloxazolidinones.
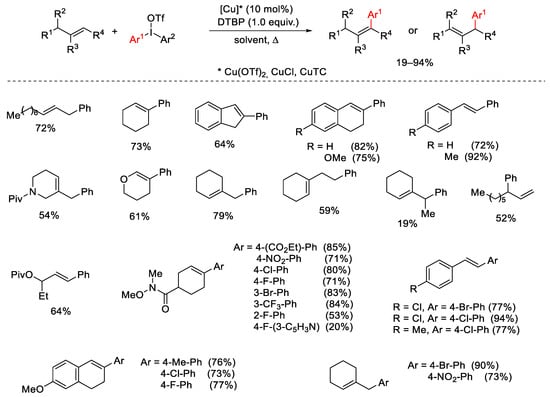
The same synthetic methodology was applied to other enolsilanes arising from enolizable carbonyls furnishing good results in enantioselective α-arylation [42] as well as to indole 3-acetamides for the construction of C3-aryl pyrroloindolines (Scheme 14) [43]. These latter can be directly prepared starting from N-tosyltryptamines by Cu(OAc)2 or Cu(OTf)2 catalyzed reactions in the presence of Ar2I⁺BF4− [44].
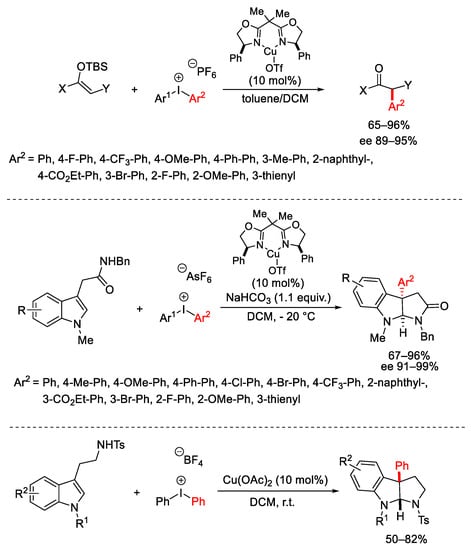
Scheme 14. α-Arylation of enolsilanes and indole 3-acetamides.
An analogous approach was applied to the total synthesis of pyrroloindoline alkaloids (+)-naseseazines A and B, exploiting the diastereoselective arylation of tryptophan derivatives 8 (Scheme 15) [45].
Scheme 15. Diastereoselective arylation of tryptophan derivatives.
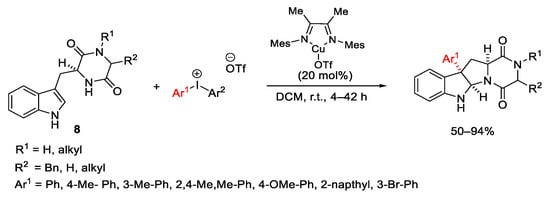
In 2012, Gaunt’s team developed a Friedel–Crafts-type procedure for the arylation of alkenes with diaryliodonium salts which behave as aromatic electrophiles (Scheme 16) [46]. This transformation can be catalyzed by Cu(I) and Cu(II) complexes and furnishes the arylation products in moderate to good yields, giving in some cases mixtures of conjugated and non-conjugated styrene products. This latter is usually the major component, and this behaviour contrasts with the outcome typically observed in the Heck conditions.
Scheme 16. Copper-catalyzed arylation of alkenes with diaryliodonium salts.
The reaction of terminal alkynes and 1-phenyl-benziodoxolones provided different types of phthalides 9 or isocoumarins 10, with a prevalence of the first, depending on the involvement of a ligand supported by the copper (Scheme 17) [47].
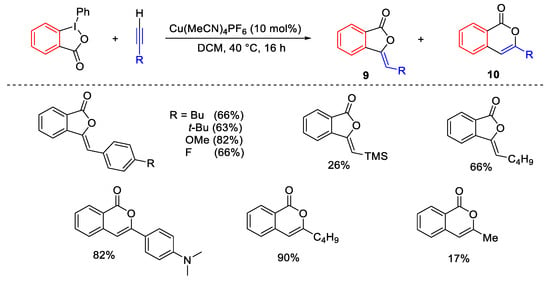
Scheme 17. Access to phthalides and isocumarins from terminal alkynes.
From the cuprate intermediate five-membered ring XIII, isocoumarin 10 will be obtained, through the intermediate XIV, if the reductive elimination involves the alkynyl ligand together with the aryl moiety (Scheme 18). If the reductive elimination takes place between the alkynyl and the alkoxy ligand, the phthalide 9 will be formed through the aryl cuprate intermediate XV, followed by intramolecular migratory insertion of the alkyne into the Ar-Cu bond as in the intermediate XVI.
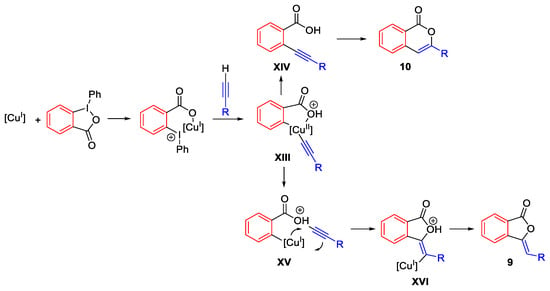
Scheme 18. Proposed mechanism for the conversion of terminal alkynes into phthalides and isocumarins.
Internal alkynes were suitable substrates for the arylation reaction with diaryliodonium salts giving α-arylketones with high regioselectivity. In this protocol, the iodonium salt acts as both as oxidant and arylation reagent (Scheme 19) [48]. The proposed mechanism considers the coordination of the electrophilic Cu(III)-aryl specie, initially formed by disproportionation of Cu(II), to alkyne.
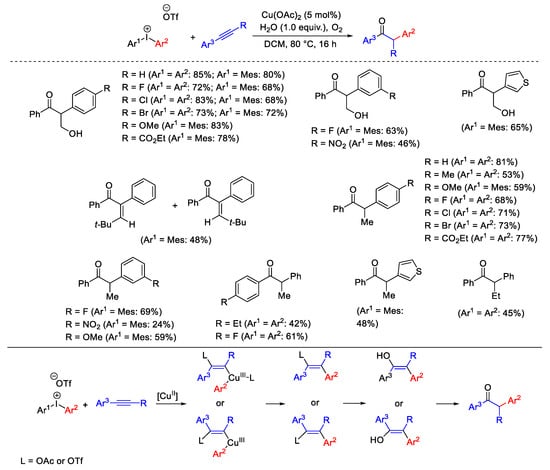
Scheme 19. Arylation reaction of internal alkynes with diaryliodonium salts to α-arylketones.
The same strategy was applied to the formation of a dihydronaphthalene scaffold, starting from arylalkynes and diaryliodonium salt in the presence of a CuCl catalyst and dtbpy as base, through the formation of a vinyl carbocation intermediate (Scheme 20) [49].
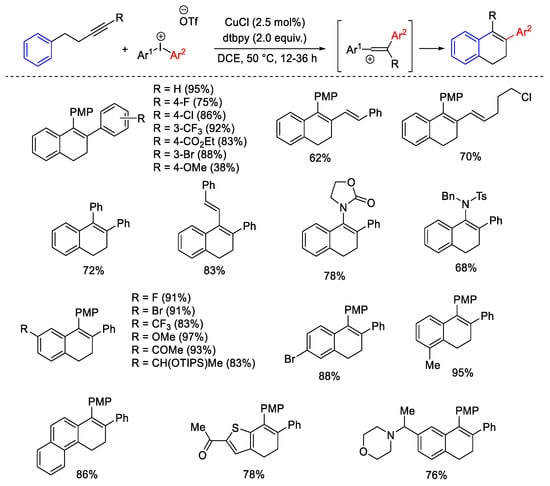
Scheme 20. Synthesis of dihydronaphthalene scaffold from arylalkynes and diaryliodonium salts in the presence of CuCl catalyst and dtbpy.
The carbocation formation was assumed also in a cascade process of alkenes (Scheme 21) and alkynes (Scheme 22) and diaryliodonium salts, with the formation of tetralins 11 or aryl cyclopentenes 12, respectively [50][51]. In both cases, the initial carbocation was moved to a remote carbon atom after the first Friedel-Crafts arylation. Concerning the first pathway, the tetralins 11 arise from the 1,2-hydride shift of the homobenzylic carbocation intermediate XVII which is formed after the initial alkene arylation process.
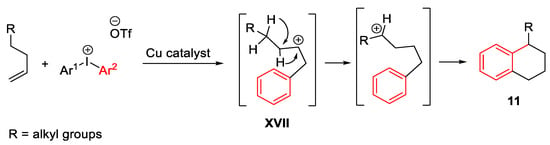
Scheme 21. Copper-catalyzed arylation of alkenes with diaryliodonium salts.
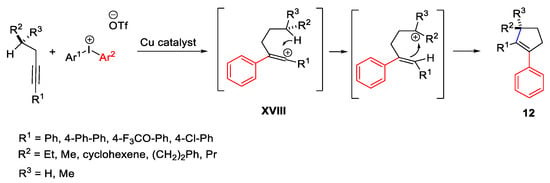
Scheme 22. Copper-catalyzed arylation of alkynes with diaryliodonium salts.
Alternatively, the formation of aryl cyclopentenes 12 took place through a concerted 1,5-hydride shift where the stereochemical information at the site of the carbocation intermediate XVIII was retained.
Alkoxyalkynes were suitable substrates to react with diaryliodonium salts affording oxygen-containing heterocycles through aryl-etherification reaction via the cleavage of a stable C–O bond (Scheme 23) [52]. The reaction occurs satisfactorily starting from aryl alkyl alkynes. Conversely, the use of diaryl alkynes [i.e., 1-methoxy-2-(phenylethynyl)benzene] provided the expected benzofuran product with only a moderate yield, due probably to the lower nucleophilicity of the phenolic oxygen. Concerning the mechanism, the diaryliodonium salt generates the electrophilic Ar-Cu(III) species which interacts with the triple bond. The so-generated vinylcarbocation XIX form the C–O bond and the final product 13 was obtained by the action of the counteranion to split the oxonium salt.
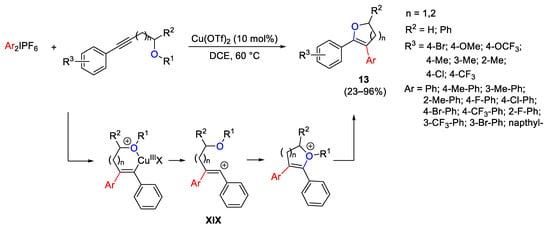
Scheme 23. Synthesis of oxygen-containing heterocycles from alkoxyalkynes and diaryliodonium salts.
Propargylamides 14 and 2-ethynylanilides 16 were used for the construction of oxazolines 15 and benzoxazines 17, respectively, bearing a tetra-substituted exocyclic double bond, through the formation of C-O and C-C bonds in a copper-catalyzed carboarylation process (Scheme 24) [53][54] The mechanism suggests the formation of Ar-Cu(OTf)2 intermediate as electrophilic species able to coordinate the triple bond and to induce ring closure via oxidative exo-dig cyclization involving the anilide oxygen as nucleophile on the internal sp carbon atom. The reaction provided a major isomer where the aryl group is in cis position to the oxygen atom of the heterocyclic ring.
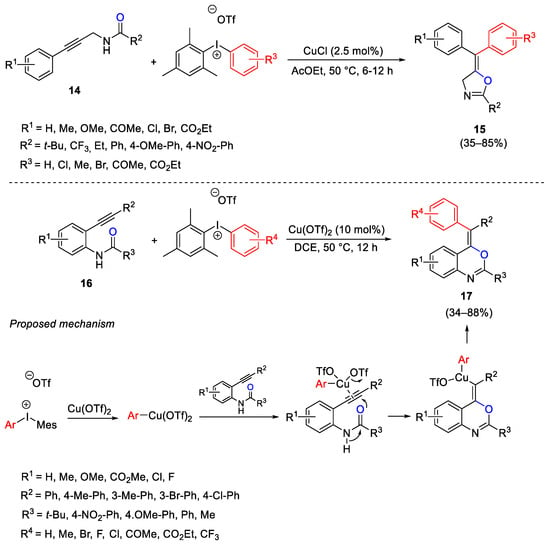
Scheme 24. Carboarylation of propargylamides and 2-ethynylanilides for the construction of oxazolines and benzoxazines.
The reactions of diaryliodonium salts in the presence of copper catalysis involving a Meyer–Schuster rearrangement of propargyl alcohols 18 afforded α-aryl α,β-unsaturated ketones 19, selectively as the E-isomers (Scheme 25). Both Cu(I) and Cu(II) can catalyze the reaction, but CuCl was identified as the optimal catalyst due to the better affinity of CuI over CuII salts for the π-orbitals of the alkynes. The reaction run on primary and tertiary propargylic alcohols and the propargylic position can be substituted with aryl, heteroaryl or olefinic groups. The methodology was applied also to ynamides, the rearrangement of which gave the corresponding arylated α,β-unsaturated imides [55].
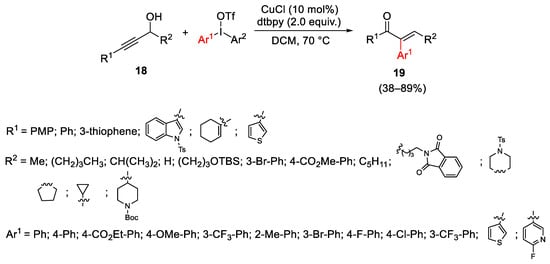
Scheme 25. Copper-catalyzed Meyer–Schuster rearrangement of propargyl alcohols.
The same authors reported the oxyarylation and oxyvinylation of allyl amides with diaryl- and vinyl(aryl)iodonium triflates providing the 5-aryl and 5-vinyl substituted oxazines 20 with excellent diastereoselectivity (Scheme 26). The mechanistic investigations to justify the stereoselection suggests a carbocation in equilibrium between syn and anti conformations prior to C-O bond formation [56].
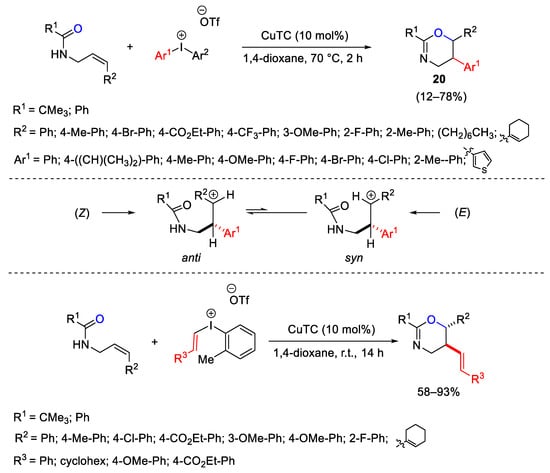
Scheme 26. Oxyarylation and oxyvinylation of allyl amides with diaryl- and vinyl(aryl)iodonium triflates.
Later, the authors observed a particular enantioselective and regiodivergent reactivity in the arylation of allylamides, dependent on the electronic properties of diaryliodonium salts (Scheme 27). In fact, the use of electron-rich diaryliodonium hexafluorophosphate, in the presence of copper(II) bisoxazoline catalyst XX and dtbpy as hindered base, afforded 1,3-diaryl oxazines 21 with excellent enantioselectivities. Then, changing the nature of the transferring aryl group from 4-methoxyphenyl to 4-trifluoromethylphenyl or other electron-deficient iodonium hexafluorophosphates, the β,β′-diaryl enamides 22 were formed with high enantioselectivity involving the shift of the double bond. It is worth noting that the allylic amides need an electron-donating alkene substituent to impart sufficient reactivity [57].
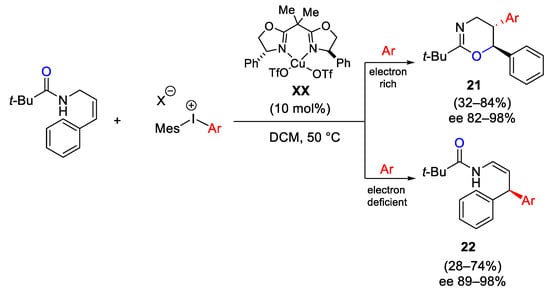
Scheme 27. Copper-catalyzed arylation of allylamides dependent on the electronic properties of diaryliodonium salts.
Cyclic nonaromatic enamides 23 showed particular behaviour as nucleophiles in direct regioselective C-3 arylation using different diaryliodonium salts and Cu(OTf)2 as a catalyst (Scheme 28) [58]. As previously reported, the Cu(III)-aryl species underwent electrophilic addition at the electron-rich C3 position, then the reductive elimination gave the final product.
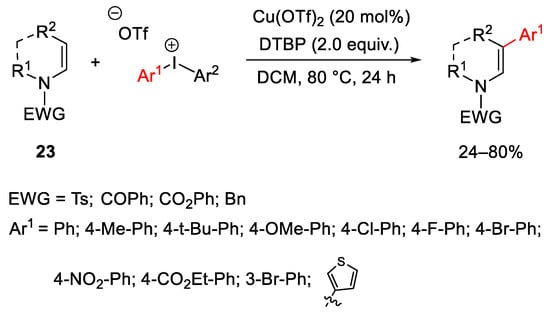
Scheme 28. Cu(OTf)2-catalyzed β-arylation of enamides.
Likewise, cyclic enamides 24 and 25 derived from tetralone and chromone, respectively, were treated with catalytic CuBr and diaryliodonium salts affording β-aryl cyclic secondary enamides 26 and 27 (Scheme 29) [59].
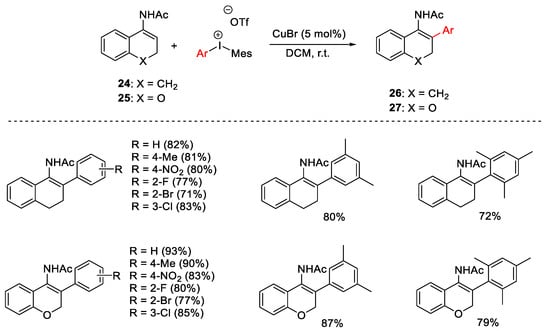
Scheme 29. β-Arylation of secondary cyclic enamides.
The reaction was extended to naphthyl-1-acetamides 28 providing C4 and C7 arylation, instead of the expected C2 aryl substituted product, due to the crucial role of the steric hindrance (Scheme 30).
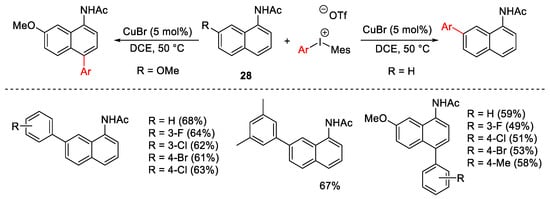
Scheme 30. C4 and C7 arylation of naphthyl-1-acetamides.
C-H arylation of different class of azaheterocycles, such as oxadiazoles, thiadiazoles, benzoxazoles and benzothiazoles was reported by employing diaryliodonium salts and a CuBr catalyst under mild reaction conditions (room temperature) or microwave irradiation (Scheme 31) [60].
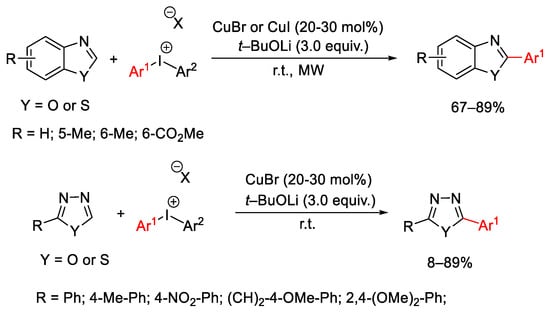
Scheme 31. C-H arylation of aza-heterocycles.
2.2. Alkynylation Reactions
Oxy-alkynylation of diazocompounds with ethynylbenziodoxolone (EBX) involving the formation of carbene intermediate was reported by the Waser group. The mild reaction conditions allowed broad applicability to diazocompounds and many functional groups are tolerated. The obtained products 29 and 30 were easily transformed into building blocks such as secondary and tertiary propargylic hydroxy acids, triazoles and isocoumarins (Scheme 32) [61].
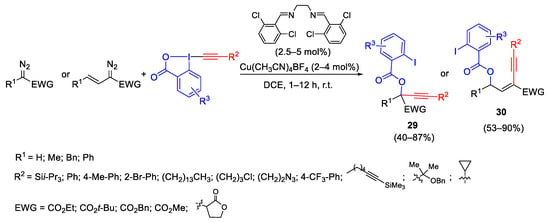
Scheme 32. Copper-catalyzed oxy-alkynylation of diazo compounds with EBX.
The proposed mechanism involves the addition of the carboxylate group of EBX to the carbene to form organocopper intermediate XXI (Scheme 33). Then, the intramolecular alkyne transfer afforded the product 29.
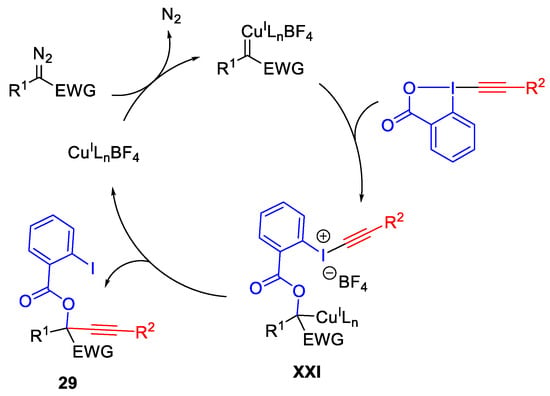
Scheme 33. Proposed mechanism for oxy-alkynylation.
Then, the same research group reported the first synthesis of ethynylbenziodazolone (EBZ) in which the oxygen atom in the iodoheterocycle was replaced by a nitrogen atom, enabling a different reactivity, applied in particular in an asymmetric oxyalkynylation of diazo compounds. The reaction was applied on a broad range of substituted diazo compounds 31 and EBZ reagents and the products 32 were obtained in high yield and enantioselectivity. The synthesis of EBZ reagent was also reported starting from the corresponding benzamides in a one-step procedure (Scheme 34) [62].
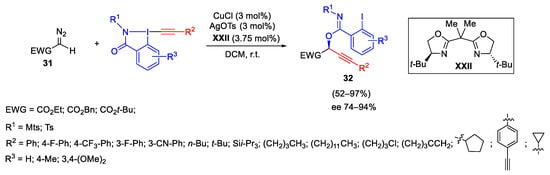
Scheme 34. Copper-catalyzed enantioselective oxyalkynylation of diazo compounds.
The oxyalkynylation reaction of the diazo compounds 33 carried out in the presence of the alcohols 34 and EBX is configured as a three-component process, leading to an extension of applicability and providing the formation of propargyl ethers 35 (Scheme 35) [63].
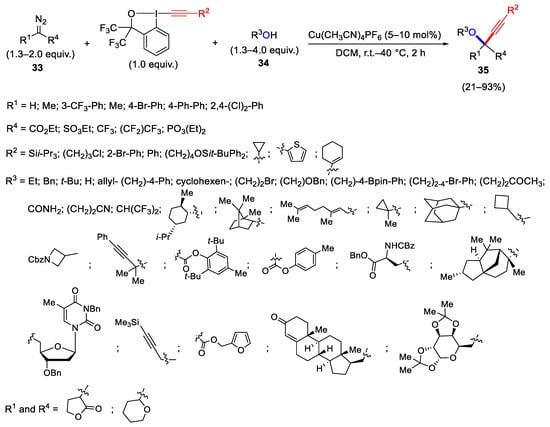
Scheme 35. Synthesis of propargyl ethers through three-component oxyalkynylation reaction.
A mechanism investigation suggested the formation of copper carbene (XXIII) and ylide intermediates (XXIV), followed by electrophilic alkynylation by EBX (Scheme 36).
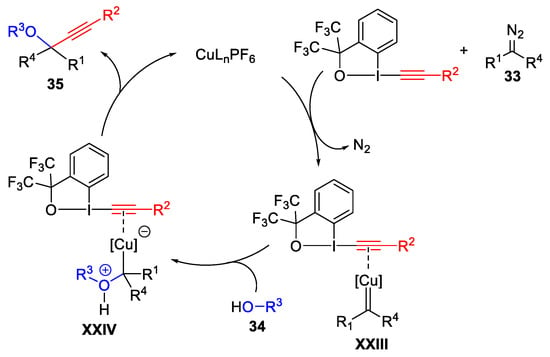
Scheme 36. Proposed mechanism for the three-component oxyalkynylation reaction.
Aminoalkynylation of terminal and internal alkenes in the presence of EBX allowed the synthesis of different azaheterocycles and the simultaneous installation of alkyne groups in one step. The mild reaction conditions allowed the use of only 1 mol% of copper catalyst. A radical intermediate was supposed for the mechanism (Scheme 37) [64].
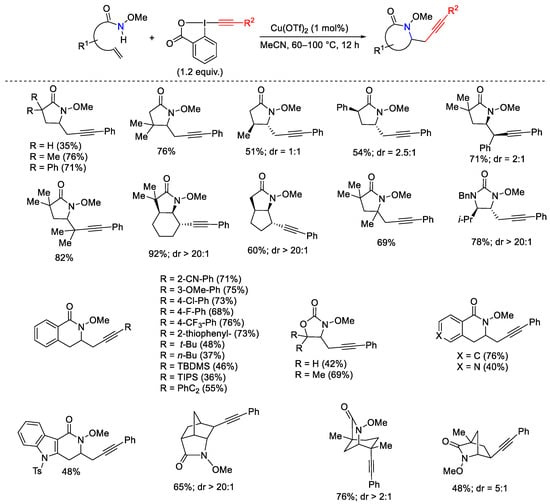
Scheme 37. Cu-catalyzed aminoalkynylation of alkenes with EBX.
The synthesis of α-alkynyl-β-substituted cyclic ketones was obtained by sequential Michael addition/electrophilic alkynylation reaction on α,β-unsaturated cyclic ketones. The reaction requires the alkyl-aluminum or Grignard reagents in the presence of copper complex as a catalyst for the first-step Michael addition, followed by electrophilic alkynylation by EBX reagent (Scheme 38) [65]. The process was extended to the coumarins affording chromanones scaffolds.
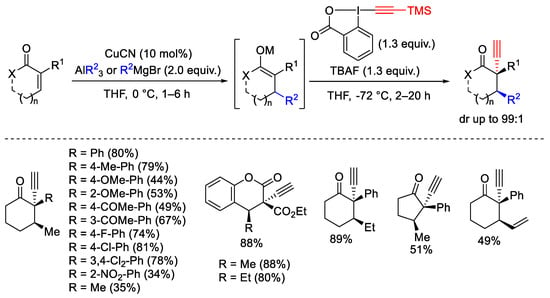
Scheme 38. Sequential Michael addition/electrophilic alkynylation on α,β-unsaturated cyclic ketones.
Brilliant results were obtained by using EBX on cyclic λ3-iodanes reporting the formation of the 4,1-benzoxazepine-2,5-diones 37 and 1,3-diynes 38. The reaction occurs by formation of the spirocyclic λ3-iodane intermediates 36, the reaction of which afforded the product by heating at high temperature, under copper catalysis and in the presence of base (Scheme 39) [66].
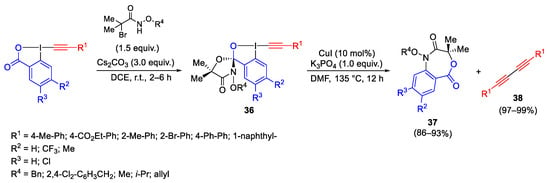
Scheme 39. Synthesis and reactivity of cyclic λ3-iodanes.
2.3. Trifluoromethylation Reactions
Among the new methods for the introduction of trifluoromethyl group into a variety of organic molecules, the use of hypervalent iodine reagents under copper catalysis was of significance for the efficiency of the reaction. Two are the useful hypervalent iodine compounds containing the CF3 group, the Togni reagents 1(I) and 2(II), reported for the first time by Togni’s research group in 2006 (Scheme 40).
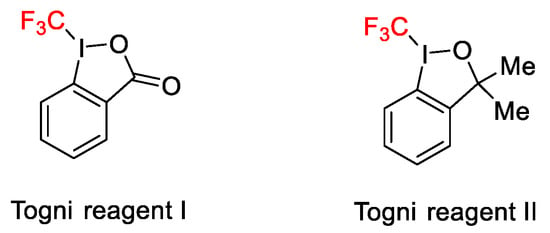
Scheme 40. Togni’s reagents.
Allylic trifluoromethylation of terminal alkenes was achieved using a cheap CuCl catalyst and Togni reagent I, the latter acting both as oxidant and the CF3 source, performing the reaction in MeOH at 70–90 °C [67]. At the same time, Buchwald obtained the same result performing the reaction with [(MeCN)4Cu]PF6 (15 mol%) catalyst in MeOH at 0 °C to r.t. (Scheme 41) [68]. The reaction tolerated a wide range of functional groups. Several mechanistic pathways could be hypothesized, but in each case, a radical species was involved.
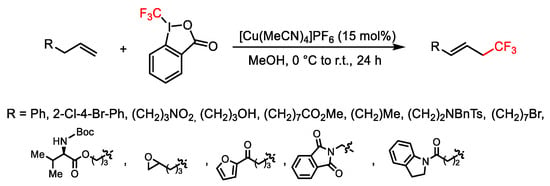
Scheme 41. Trifluoromethylation of terminal alkenes.
Hydrofunctionalization of homoallylic alcohol derivatives, with anti-Markovnikov regioselectivity, was achieved using hypervalent iodine(III) reagent containing the CF3 (Togni reagent I) or N3 source. The benzyl group present on the substrate was essential, playing the role of redox-active hydrogen donor. In these conditions several functional groups were tolerated [69]. The mechanistic pathways suggested the formation of the CF3 radical followed by a 1,5-H radical shift affording the benzyl radical then oxidized to the oxonium cation XXV. The subsequent reaction with MeOH produced the acetal 39 hydrolysable in the presence of TsOH. The method was also applied to the 5-hexenol and 6-heptenol derivatives, in the presence of catalyst Cu(OAc)2, resulting in the formation of the trifluoromethylated ketones (Scheme 42).
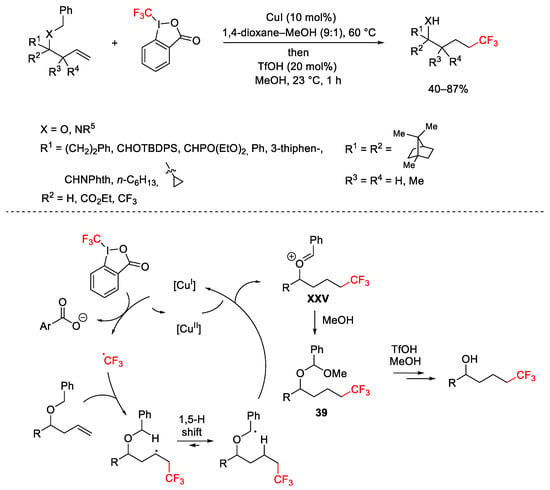
Scheme 42. Trifluoromethylation of homoallylic alcohol derivatives.
The protocol was not limited to the introduction of a CF3 group, but using azidobenziodoxolone (ABX, Zhdankin reagent) as a source of the azido radical, the formation of azidoalcohols 40 was reported as hydroazidation of homoallylic alcohols (Scheme 43).
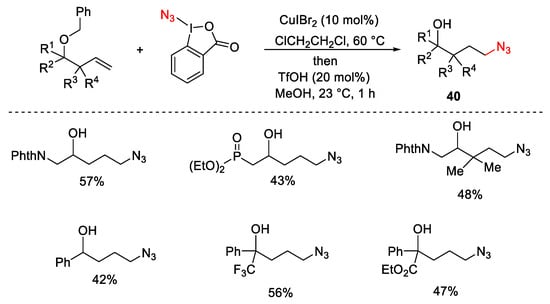
Scheme 43. Formation of azidoalcohols.
The trifluoromethylation of allylsilanes was reported with Togni reagent I. The reaction on 2-substituted allylsilanes 41 gave desilylated product 42 bearing the trifluoromethyl group in the allylic position, while the reaction of unsubstituted allylsilanes afforded the corresponding vinyl silane derivatives 43 (Scheme 44) [70].
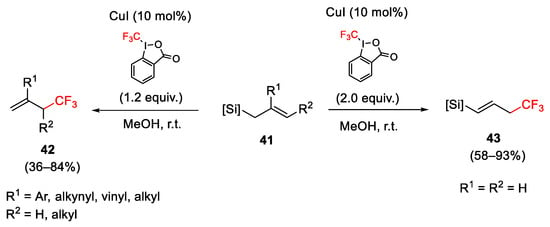
Scheme 44. Trifluoromethylation of allylsilanes with Togni reagent I.
The copper-catalyzed trifluoromethylation of substituted stirenes, with Togni reagent II and in the presence of DBU, provided trifluoromethylated derivatives with total regioselectivity (Scheme 45) [71].
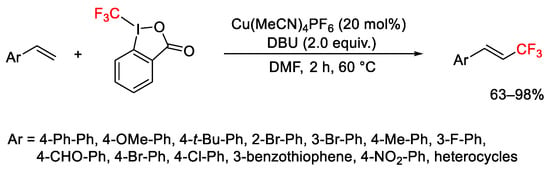
Scheme 45. Trifluoromethylation of styrenes.
Additionally, terminal alkynes were subjected to the trifluoromethylation process affording substituted acetylenes [72]. The mild reaction conditions allowed tolerance of different functional groups. The presence of a ligand, such as the 1,10-phenanthroline, gave the complexation with the copper allowing the formation of the copper-acetylide species XXVI favoured by the base KHCO3 (Scheme 46). The subsequent oxidative addition of CF3 led to complex XXVII and the final reductive elimination of copper afforded the product and regenerated the copper(I).
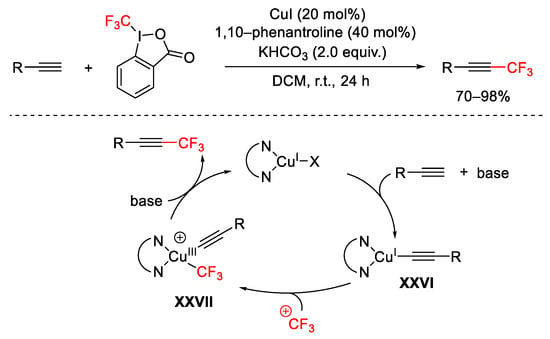
Scheme 46. Trifluoromethylation of terminal alkynes.
Trifluoromethylation of electron-rich enamides 44 was realized with Cu(MeCN)4PF6 as catalyst and Togni reagent I in THF at r.t. affording 45 with E stereoselectivity (Scheme 47) [73]. The authors suggested the formation of an α-trifluoromethyl imine intermediate XXVIII, which could react following two divergent routes, depending on the solvent and catalyst used. When the reaction was performed with CuCl as catalyst in MeOH, the oxytrifluoromethylation process gave selectively product 46.
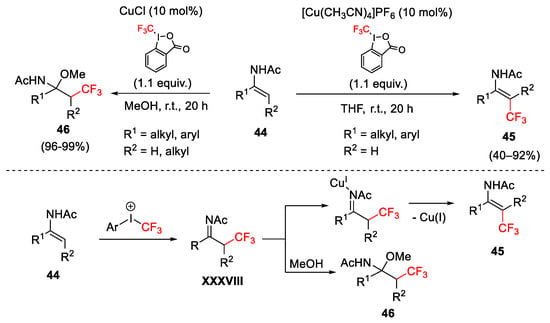
Scheme 47. Trifluoromethylation of electron-rich enamides.
When the amide acted as electron-withdrawing group, the trifluoromethylation on the double bond required more drastic reaction conditions and resulted in Z stereoselectivity, due to the action of the amide group as directing-group (Scheme 48) [74].
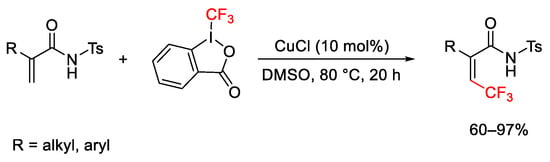
Scheme 48. Trifluoromethylation of electron-deficient alkenes.
The method was extended to other α,β-unsaturated carbonyl compounds including enones, esters, and thioesters, providing (E)-α-trifluoromethylated products 47 in good yields, with total regio- and stereoselectivity (Scheme 49) [75].
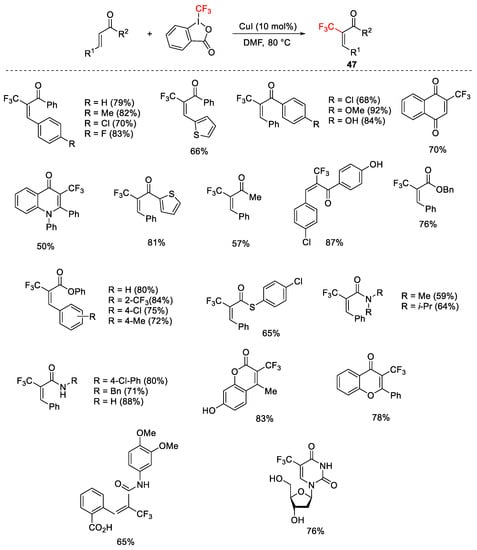
Scheme 49. Trifluoromethylation of α,β-unsaturated carbonyl compounds.
An experiment carried out to shed light on the reaction mechanism indicated clearly that the reaction proceeded via a radical process.
The CF3 radical intermediate was also involved in the direct C-H trifluoromethylation of quinones. The mild reaction conditions were compatible with different substituents on the substrates (Scheme 50) [76].
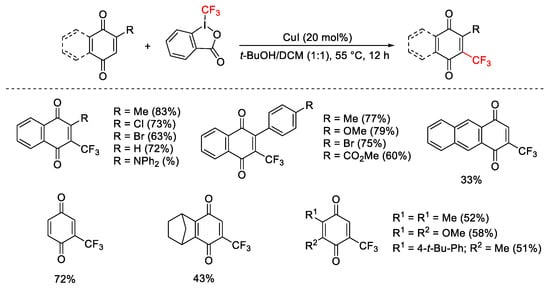
Scheme 50. Trifluoromethylation of quinones.
Enantioselective trifluoromethylation of cyclic β-ketoesters was obtained by performing the reaction with Togni reagent II, Cu(OTf)2 catalyst, in the presence of boxmi ligand XXIX [77]. Five-membered rings were converted to the corresponding trifluoromethylated derivatives 48 in high yields and ee up to 99% (Scheme 51). Conversely, the 5-(trifluoromethyl)dibenzothiophenium tetrafluoroborate (Umemoto’s reagent) was used as trifluoromethyl source for the functionalization of six-membered rings.
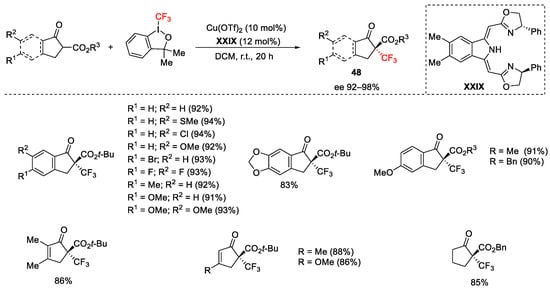
Scheme 51. Trifluoromethylation of β-ketoesters.
The use of Togni reagent II in the presence of chiral organocatalyst imidazolidinone XXX allowed the enantioselective trifluoromethylation of aldehydes (Scheme 52). The suggested mechanism excluded the involvement of a radical pathway but involved the formation of the chiral enamine between the aldehyde and the imidazolidinone followed by the formation of the λ3-iodane XXXI species able to give the C-CF3 bond formation [78].
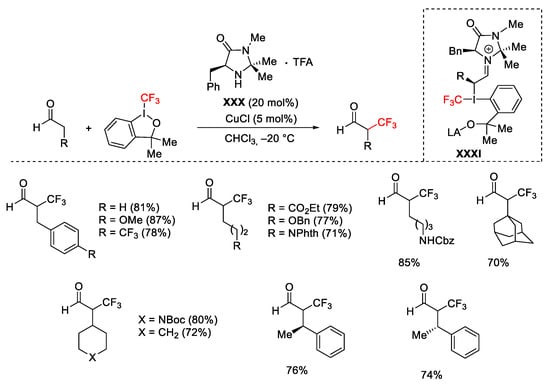
Scheme 52. Enantioselective α-trifluoromethylation with organocatalyst.
Monteiro’s group reported the trifluoromethylation reaction of N,N-dialkylhydrazones derived from aromatic aldehydes and α,β-unsaturated aldehydes. The reactions were performed with Togni reagent I, CuCl catalyst in chloroform as solvent, at r.t., affording trifluoromethylated hydrazones and β-trifluoromethylated alkenylhydrazones (Scheme 53) [79][80].
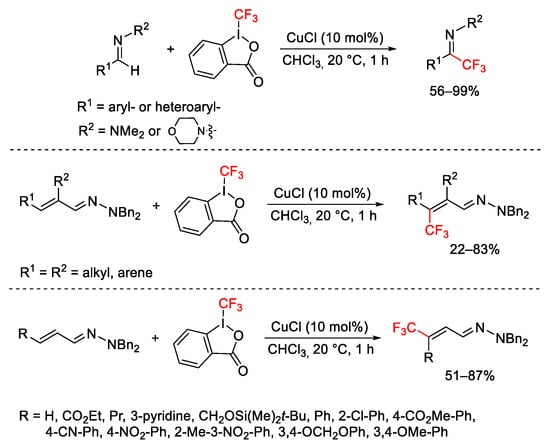
Scheme 53. Copper-catalyzed trifluoromethylation of hydrazones.
By performing the reaction on the N-monosubstituted hydrazones arising from ketones, quaternary α-trifluromethyl hydrazines 49 were isolated (Scheme 54) [81].
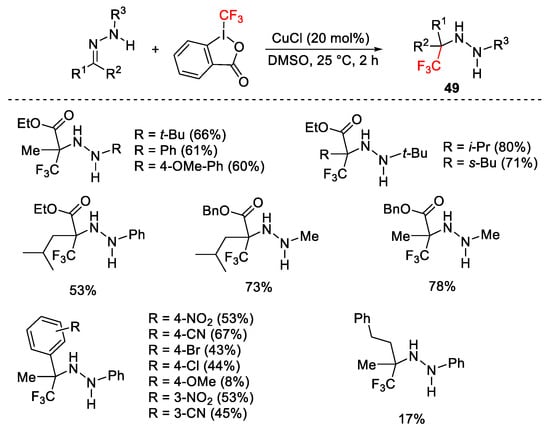
Scheme 54. Trifluoromethylation of N-monosubstituted hydrazones.
In both cases, the CF3 radical is generated through a single electron transfer promoted by the copper salt affording intermediate XXXII (Scheme 55). At this point, this latter was oxidized by the copper(II) salt affording the cationic species XXXIII which generate the previously described trifluoromethylated compounds.
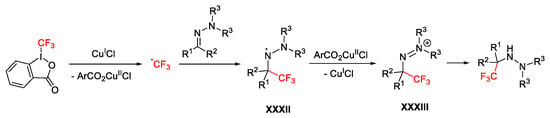
Scheme 55. Mechanism for the trifluoromethylation of hydrazones.
Cycloalkanols reacted with hypervalent iodine reagents affording terminal trifluoromethylated ketones via selective C-C cleavage of cycloalkanols. The Togni reagent facilitated the oxidation of the substrate to the alkoxy radical, the β-fragmentation of which produced the ketone-bearing terminal alkyl radical able to afford trifluoromethylation adduct (Scheme 56) [82].
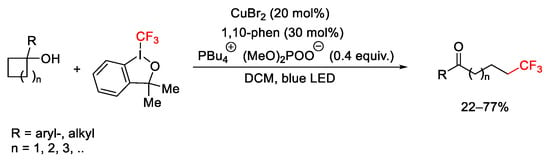
Scheme 56. Terminal trifluoromethylation of ketones by C-C bond cleavage of cycloalkanols.
Trifluoromethylation of propargylic secondary and tertiary alcohols afforded α-substituted enones 50 via Meyer–Schuster rearrangement (Scheme 57) [83].
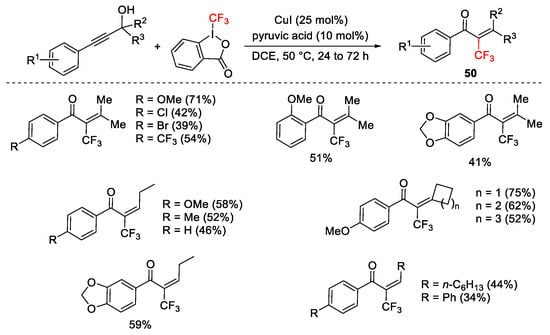
Scheme 57. Copper-catalyzed domino trifluoromethylation/Meyer–Schuster rearrangement.
This approach was applied also for a one-pot procedure to access CF3-isoxazole and CF3-pyrazole derivatives subjecting the propargyl alcohol in the presence of Togni’s reagent I and hydroxylamine and hydrazine, respectively (Scheme 58).
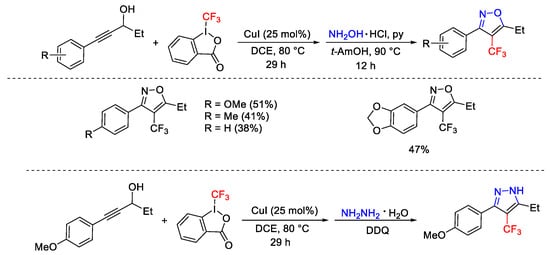
Scheme 58. Copper-catalyzed domino trifluoromethylation/Meyer–Schuster rearrangement.
Regioselective ortho trifluoromethylation of arenes and heteroarenes by using the pivalamido substituent as the directing group was reported in t-BuOH as solvent, with CuCl and Togni reagent I (Scheme 59) [84].
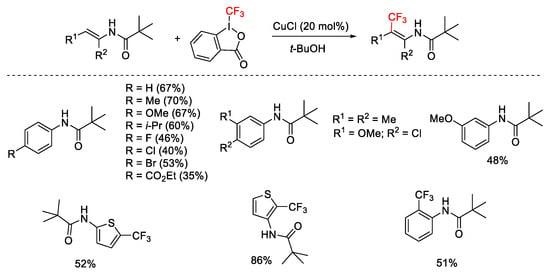
Scheme 59. Copper-catalyzed trifluoromethylation of (hetero)arenes with Piv as directing group.
Trifluoromethylation of aryl and vinyl boronic acids [85] and organotrifluoroborates [86] were obtained by using different copper catalysts, ligands, and solvents, but always under mild reaction conditions (Scheme 60).
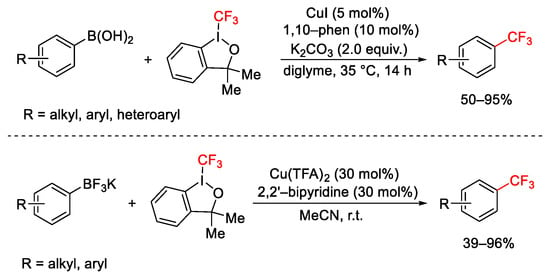
Scheme 60. Trifluoromethylation of organo boronic acids and organotrifluoroborates.
Trifluoromethylated acetylenes 51 were also obtained starting from alkynyltrifluoroborates, by using CuSCN catalyst and 2,2′-bipyridine ligands in MeCN at r.t. (Scheme 61) [87].
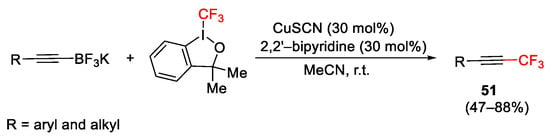
Scheme 61. Trifluoromethylation of alkynyltrifluoroborates.
Trifluoromethylation of various C3-substituted indole derivatives was realized under mild conditions, with 10 mol% of CuOAc and Togni reagent I in MeOH, obtaining C2-trifluoromethylated indoles (Scheme 62) [88]. The regioselectivity was obtained also on substrates not substituted in position 3. The presence of the electron-withdrawing group at N1 position influenced in a negative way the reaction, resulting in low yield.
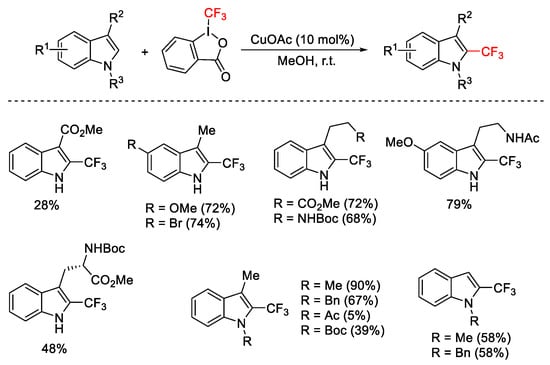
Scheme 62. Trifluoromethylation of C2 position of indoles.
Trifluoromethylation was also involved in difunctionalization processes as aminotrifluoromethylation, oxytrifluoromethylation, and cianotrifluoromethylation.
Terminal alkenes and alkynes were suitable substrates to give trifluoromethyl-benzoyloxylation in the presence of Togni reagent I and copper(I) catalyst (Scheme 63). The reaction proceeded with high regio- and stereoselectivity. The alkenes reacted faster than alkynes and the presence of electron-donating substituents accelerated the reaction [89][90].
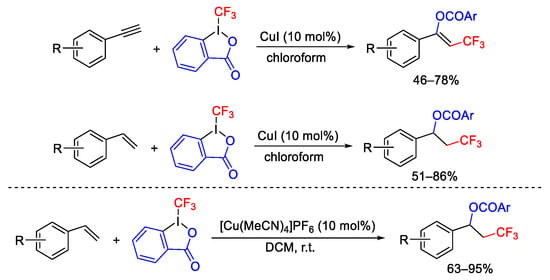
Scheme 63. Oxytrifluoromethylation of alkenes and alkynes.
Tandem oxymonofluoromethylation of alkenes was developed merging the iodine(III) reagent containing monofluoroacetoxy ligand 52, acting as bifunctional reagent, as fluoromethyl and oxygen source and the visible-light copper(I) photoredox catalysis able to generate the fluoromethyl radical (Scheme 64) [91].
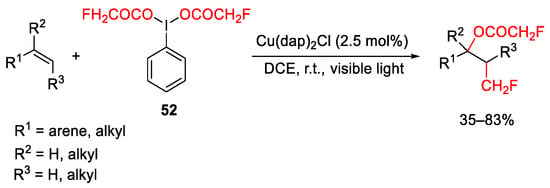
Scheme 64. Copper-catalyzed oxy-monofluoromethylation of alkenes.
Trifluoromethylazidation of alkenes was obtained by using Togni reagent II in the presence of TMSN3 as a second nucleophile under Cu(CH3CN)4PF6 as catalyst [92]. In this way, a variety of CF3-containing organoazides was synthetized starting from different kinds of olefins (Scheme 65).
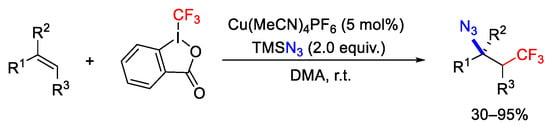
Scheme 65. Copper mediated trifluoromethylazidation of alkenes.
Different aryl-substituted styrenes could undergo a cyanotrifluoromethylation with Togni reagent I and in the presence of a stoichiometric amount of CuCN [93].
The reactions of the alkenylamines under copper catalysis and in the presence of Togni reagent provided different results depending on the reaction conditions [94]. Performing the reaction in t-BuOH, N-migratory oxytrifluromethylation was observed to furnish compounds 53 (Scheme 66). This result proceeded via an aziridine intermediate, also isolated during the reaction. Aziridine 54 was selectively formed performing the reaction in DCM. Then, the formed aziridine, in the presence of a nucleophile provided a second step with the addition of the second nucleophile.
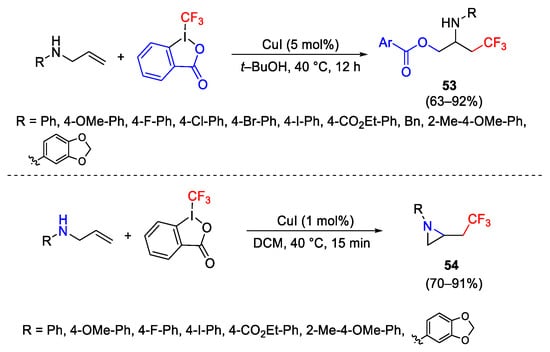
Scheme 66. Synthesis of β-trifluoromethylamines.
Aminotrifluoromethylation of alkenylamines was achieved with Togni reagent and catalytic CuI, resulting in the formation of trifluoromethylated cyclic amines [95]. In the case of trifluoromethylated pyrrolidines 55, the presence of triethylamine as co-catalyst improved the yield trapping the 2-iodobenzoic acid and avoided the protonation of the substrate (Scheme 67). The key intermediate was the formation of trifluoromethyl radical. Mechanistic studies revealed the positive effect of the pretreatment of the Togni reagent with CuI with the formation of the reactive species Cu(II), generated by oxidation, that served as a Lewis acid catalyst to activate Togni reagent enhancing the electrophilicity of the hypervalent iodine.
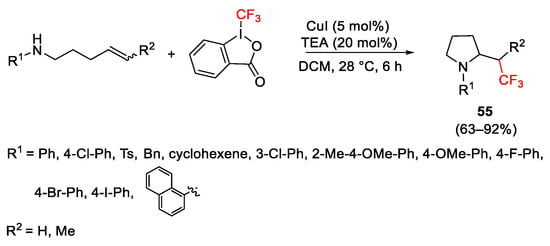
Scheme 67. Synthesis of β-trifluoromethylamines.
2-Vinylbenzamides and other terminal alkenes bearing an amide functional group as nucleophile gave functionalized lactames exploiting the aminotrifluoromethylation difunctionalization domino process (Scheme 68) [96].
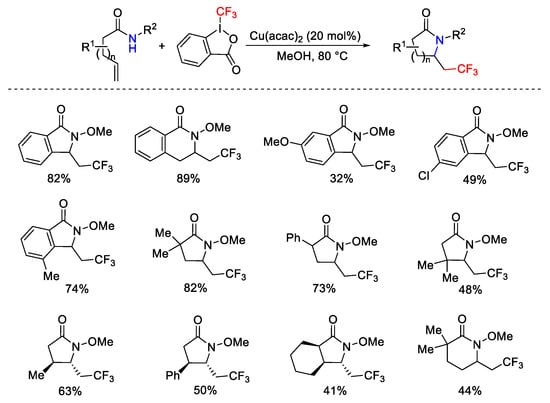
Scheme 68. Synthesis of CF3-containing lactams.
The [(MeCN)4Cu]PF6 (15 mol%) catalyst was exploited on unactivated alkenes bearing a -OH group as a nucleophilic pendant resulting in the formation of β,γ,δ-lactones, cyclic ethers, and epoxides through the oxytrifluoromethylation process (Scheme 69) [97]. In addition to copper catalyst, the presence of a pyridine-based bidentate ligand, as the 2,2′-biquinoline, was essential to avoid metal elimination and to observe the difunctionalization process. The complete inhibition of the reaction in the presence of TEMPO suggested a radical mechanism for the process.
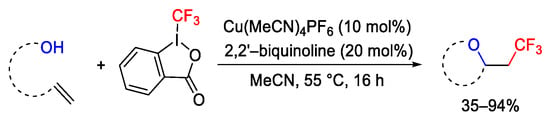
Scheme 69. Oxytrifluoromethylation of unactivated alkenes.
The trifluoromethylation of unsaturated-carboxylic acids in the presence of chiral ligands such as (S,S)-t-BuBox afforded lactones 56 through the enantioselective oxytrifluoromethylation process (Scheme 70) [98].
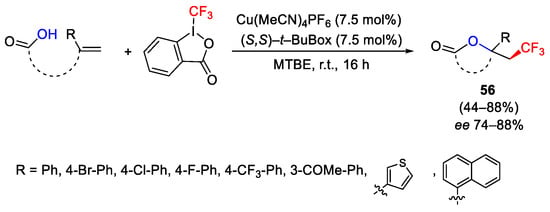
Scheme 70. Asymmetric oxytrifluoromethylation of alkenes.
A diastereoselective 1,4-difunctionalization of 1,3-dienes 57 was obtained by treatment with CuCN/t-Bu3P catalytic system. The presence of bulky phosphine accelerated the reaction affording acyloxytrifluoromethylation products 58 with the regioselective addition of the 2-iodobenzoate to the more sterically hindered carbon atom (Scheme 71) [99].
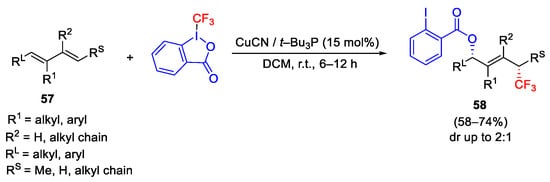
Scheme 71. Copper-catalyzed acyloxytrifluoromethylation of dienes.
Oxytrifluoromethylation of the 2,3-allenoic acids 59 provided β-trifluuoromethylated butenolides 60 in the presence of CuBr as catalyst and 1,10-phenthroline-5,6-dione as ligand, in dichloromethane under mild reaction conditions (Scheme 72) [100].
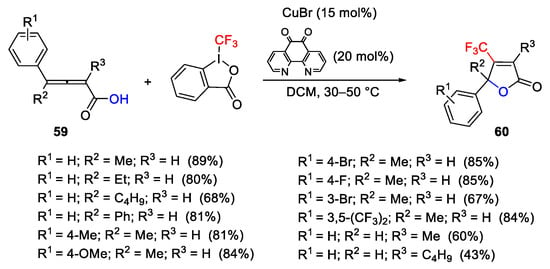
Scheme 72. Copper-catalyzed oxytrifluoromethylation of 2,3-allenoic acids.
Heterocycles as 5-trifluoroethyl isoxazolines were obtained by intramolecular reaction of β,γ-unsaturated oximes with Togni reagent I and CuCl in the presence of base (Scheme 73) [101].
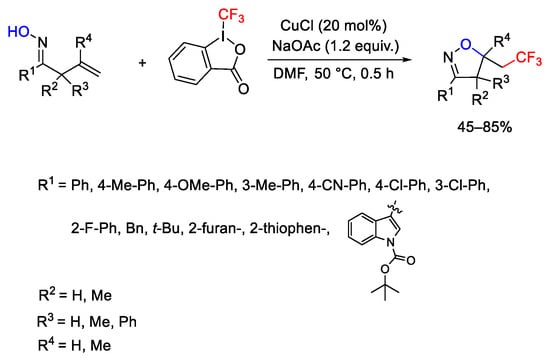
Scheme 73. Copper-catalyzed synthesis of trifluoromethyl-substituted isoxazolines.
Carbocycles were obtained by trifluoromethylation-cyclization of simple 1,6-enynes using a C-C triple bond as nucleophile. The reaction was totally regioselective affording five- and six-membered rings in good yields (Scheme 74) [102].
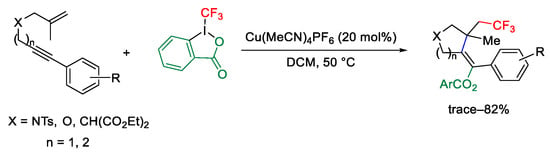
Scheme 74. Copper-catalyzed trifluoromethylation/cyclization of enynes.
Carbocycles and aza-heterocycles were synthesized exploiting the intramolecular carbotrifluoromethylation and aminotrifluoromethylation processes using the Cu(I)/Togni system [103]. Starting from simple alkenes bearing allylic protons, the choice of the solvent was important in order to avoid competitive deprotonative trifluoromethylation reaction (Scheme 75). The use of N-allylanilines and homoallylanilines afforded the corresponding five- and six-membered heterocycles.
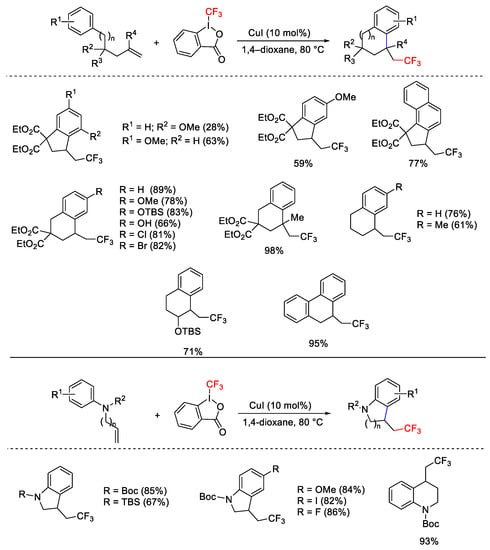
Scheme 75. Domino C-C and C-CF3 bond formations.
Starting from alkenyl amines and 2-allyl anilines, the synthetic path provided the 2-trifluoroethyl-substituted pyrrolidines and indolines [104]. The mild conditions allowed the reaction to be performed in the presence of different functional groups (Scheme 76).
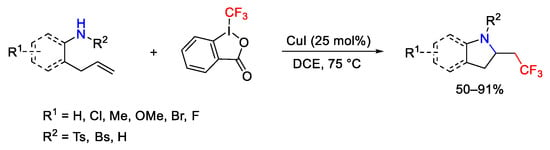
Scheme 76. Aminotrifluoromethylation of unactivated alkenes.
3. Hypervalent Iodine as Reagent in C-N Bond Formation
3.1. Arylation Reactions
Diaryliodonium salts have been employed as arylating agents of a wide range of nitrogen nucleophiles under copper(I)-catalyzed conditions.
The most general and accepted mechanism, depicted in Scheme 77, occurs through oxidative addition of the diaryliodonium salt with the Cu(I)-catalyst forming the electrophilic Cu(III)-intermediate XXXIV. Coordination of the nitrogen nucleophile generates XXXV which in turn, in the presence of a base, undergoes reductive elimination giving the target product with regeneration of the Cu(I)-catalyst.
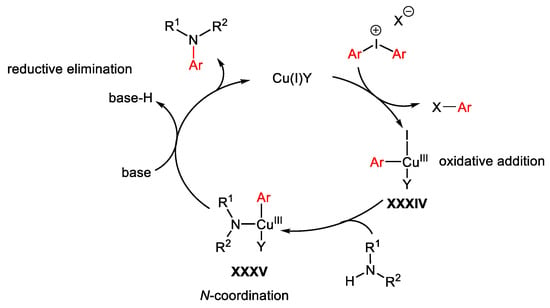
Scheme 77. General mechanism for Cu-catalyzed C-H amination reactions in the presence of hypervalent iodine reagents.
Among the copper(I) sources, copper halides are the most widely exploited to catalyze N-arylation reactions using diaryliodonium salts. Two of the first reported cases date back to 2002, when the Chen research group used CuI to promote the N-arylation of indole and benzimidazole with diaryliodonium tetrafluoroborates bearing various substituents in para (i.e., methyl, chloro, methoxy, bromo, and acetamido) or meta (nitro) positions (Scheme 78) [105][106]. The reaction is general for these substrates and proceeds in good yields using potassium carbonate as the base and DMF as the reaction solvent.
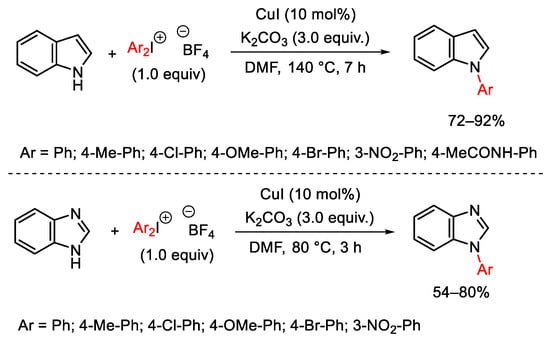
Scheme 78. Copper(I)-catalyzed arylation of indole and benzimidazole.
Chen and co-workers took advantage of this protocol to promote N-arylation of tetrazole and uracil compounds (Scheme 79) [107][108]. In the latter case, the reaction furnished the N,N-diarylation products 61.
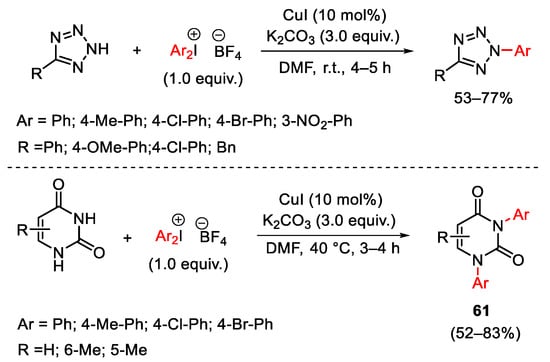
Scheme 79. Copper(I)-catalyzed arylation of tetrazole and uracil derivatives.
Conversely, when the pyrimidine reactivity was investigated in the presence of NaOAc instead of K2CO3, mono arylation occurred. In particular, 6-methyluracile led to the 3-phenyl-substituted product 62, while thymine and uracil showed selectivity at position 1, giving 63 (Scheme 80). The authors attribute the regioselectivity to steric and acidic factors.
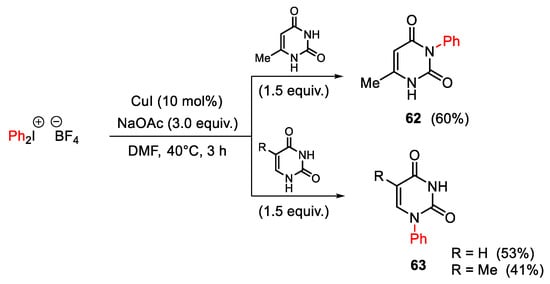
Scheme 80. Regioselective copper(I)-catalyzed arylation of 6-methyluracile, thymine and uracil.
Copper(I) iodide was the catalyst of choice in the work by Thakur and co-workers for the selective N3-arylations of hydantoins with diphenyliodonium triflate as the aryl partner (Scheme 81) [109]. K3PO4 and DCE were proven to be the best base and solvent. This approach works with a wide range of hypervalent iodine(III) salts containing bulky, electron-neutral, electron-donating or electron-withdrawing groups. Congested hydantoins were also well tolerated. Finally, the N3 mono-arylated products can be arylated at the N1-position under the same reaction conditions by operating in dioxane instead of DCE.
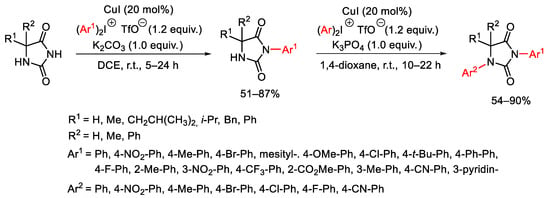
Scheme 81. Copper(I)-catalyzed arylation of hydantoins.
CuCl was employed as catalyst for the synthesis of N-arylsulfonamides through an environment-friendly procedure in which a series of diaryliodonium triflates and N-H sulfonamides were reacted in the presence of K3PO4 using water as the reaction medium (Scheme 82) [110]. In particular, the unsymmetrical 4-methoxy-4′-nitro diphenyliodonium salt transferred the electron deficient aryl ring, while the unsymmetrical mesityl-phenyliodonium salt selectively furnished the less hindered arylsulfonamide.
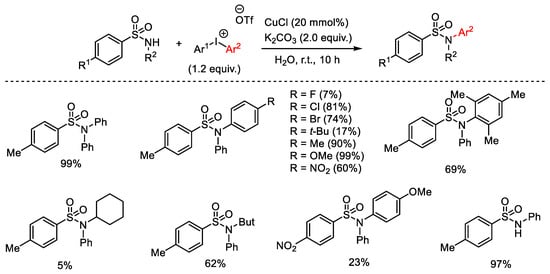
Scheme 82. Copper(I)-catalyzed arylation of sulfonamides with diaryliodonium salts.
A sustainable C-N cross-coupling reaction promoted by CuBr using hypervalent iodine salts was reported by Varma in 2012 [111]. The reaction involves an ultrasound accelerated N-arylation of sulfoximines carried out in the presence of CuBr catalyst, diaryliodoniumtriflate as electrophilic agent in aqueous polyethylene glycol as the benign reaction solvent (Scheme 83). It is worth noting that ultrasonication dramatically improved the yield of products 64 while, the addition of a base did not influence the reaction.
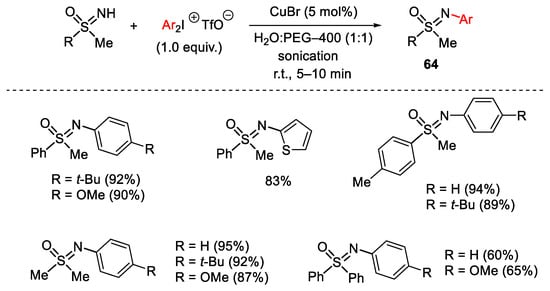
Scheme 83. Copper(I)-catalyzed arylation of sulfoximines with diaryliodonium salts.
Moryiama group developed the synthesis of 3-bis-sulfonimido-indole derivatives using indolyl(aryl)iodonium imides. The process proceeded through the C-N bond formation between nucleophilic indoles and nucleophilic amides. Among different ligands, the presence of 2-butoxy group on the aryl induced high indole selectivity by a steric effect (Scheme 84) [112][113].
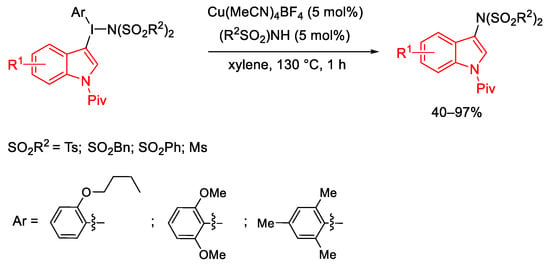
Scheme 84. Copper(I)-catalyzed C-N coupling reaction using indolyl(aryl)iodonium imides.
To avoid the formation of in situ generated sulfonylimino-λ3-iodanes, the research group of Suna develop a one-pot two-step strategy for the direct C-H amination of indoles and other heteroarenes working with a copper(I) source (Cu(MeCN)4BF4) and an unsymmetrical iodonium salt (Scheme 85) [114]. This one-pot method was developed also for the para-selective C-H amination of carbocyclic arenes with a number of N-unprotected amines [115]. The regioselective process is a result of the combined directing effects of arene substituents typically strongest electron-releasing substituents.
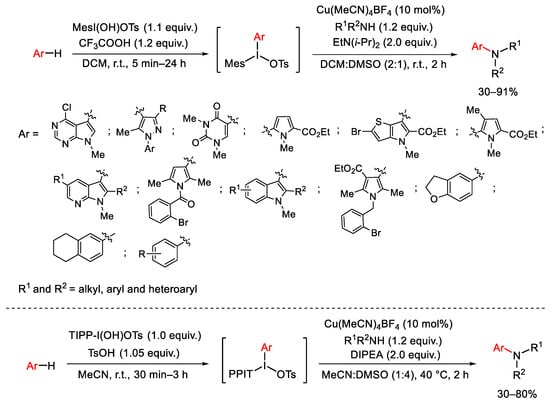
Scheme 85. Copper(I)-catalyzed C-H amination of unsymmetric diaryliodonium salts with amine compounds.
On these bases, Shafir and co-workers developed a procedure for the selective synthesis of N-aryl-5-iodoimidazoles by copper-catalyzed intramolecular aryl migration of a new family of λ3-iodanes 65, employing Cu(OTf)2 as pre-catalyst (Scheme 86). These conditions tolerate electron-donating and mild electron-withdrawing substituents on the aryl fragment [116]. The highest yields and selectivity toward the 1,5-disubstituted products were achieved using N-Me-benzimidazole as an additive able to be included in the coordination sphere of copper and thus enforcing the intramolecular phenyl transfer.
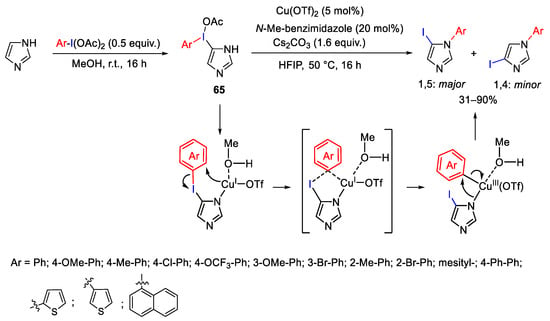
Scheme 86. Copper catalyzed aryl migration: selective synthesis of N-aryl-5-iodoimidazoles.
Diaryliodonium salts under copper catalysis are also prominent reagents for the aryl quaternization of N-heteroarenes without N–H groups. Gao and his group proposed a procedure for the direct aryl quaternization of N-substituted imidazole derivatives using Cu(OAc)2 as catalyst and a series of diaryl iodonium salts bearing either electron-deficient or electron-rich aryl moieties (Scheme 87) [117]. Because radical scavengers did not influence the reaction course, the process plausibly involves the classic mechanism for Cu-catalyzed C–H amination by diaryliodonium salts through a Cu(III) complex.
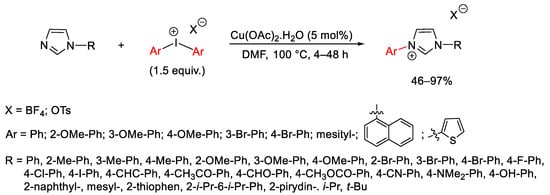
Scheme 87. Aryl quaternization of imidazole derivatives under copper catalysis.
A strictly related strategy was explored by Baumgartner for the single-step preparation of N,N′- diarylated phosphaviologens (Scheme 88) [118]. This method was proven to be compatible with both electron-poor and electron-rich diaryliodonium salts, even though low yields of products 66 were isolated when sterically demanding iodine(III) compounds were employed. It is worth noting that compounds 66 could be employed as electroactive and electrochromic materials.
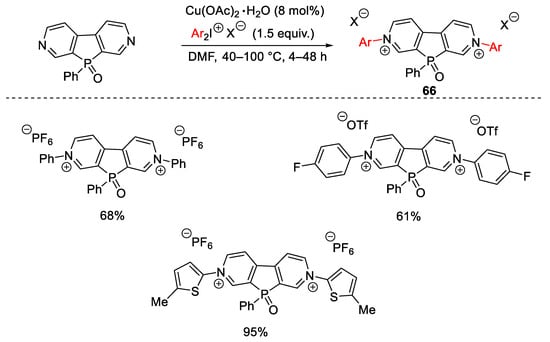
Scheme 88. Aryl quaternization of electron-deficient pyridines under copper catalysis.
Activation of nitrile groups via the intermediacy of Cu(III)-species with hypervalent iodine compounds have also been realized. Cheng reported a one-pot approach to substituted ureas 67 from N-phenylcyanamide (Scheme 89) [119]. The base-promoted N-arylation reaction toward the formation of XXXVI, is followed by the addition of Cu(OTf)2, a slight excess of a second diaryliodonium salt and AcOH. The second reaction step involves the conversion of Cu(OTf)2 into Cu(I)OTf by either reduction or disproportion. Oxidative addition to the Cu(I)-cation by the diaryliodonium salt generates the Ar2−Cu(III)-species, which acts as carbocation equivalent and, in turn, is able to transfer the aryl moiety to the cyano functional group of XXXVI generating XXXVII and restoring the catalyst. The target product 67 was formed by hydration of the intermediate XXXVII.
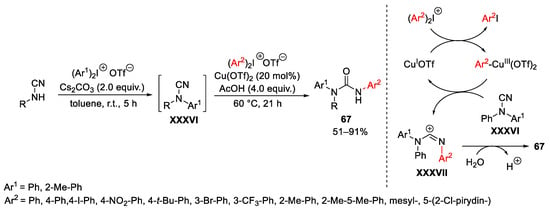
Scheme 89. Copper-catalyzed synthesis of unsymmetrical diarylureas.
The reactivity of diaryl hypervalent iodonium reagents toward nitriles was extended to the related cyclic iodonium salts. In this way, Chen et al. synthesized a series of diarylmethane amide compounds [120]. As depicted in Scheme 90, the transformation occurred by reacting cyclic diaryliodonium salts with nitriles using Cu(I)Cl as the catalyst. Furthermore, the desired amides were isolated in good yields independent from the electron property of the substituents on the diphenyl iodonium.
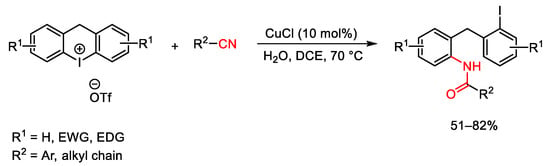
Scheme 90. Copper-catalyzed synthesis of diarylmethane amides using cyclic iodonium salts.
The activation ability by Cu(III)-aryl species toward nitriles was exploited for the construction of heterocyclic skeletons. In particular, Novak described an oxidative arylation–cyclization procedure for the construction of iminobenzoxazines 68 from ortho-cyanoanilides and unsymmetric diaryliodonium triflates bearing a bulky mesityl residue as the non-transferable aromatic ring (Scheme 91) [121]. The process showed fairly good functional group tolerance. Concerning the reaction mechanism, the authors suppose that the electrophilic Cu(III)-specie is able to react with the nitrile residue of the starting compound leading the cationic specie XXXVIII and XXXVIII′, restoring the Cu(I) catalyst. Cations XXXVIII generate 68 through an intramolecular 6-exo-dig cyclisation reaction.
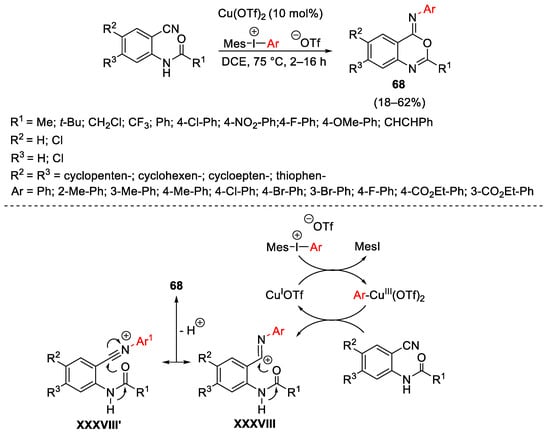
Scheme 91. Copper-catalyzed ring closure of o-cyanoanilides with hypervalent iodine reagents.
Along with this type of reactivity, Chen and co-worker proposed a regioselective copper-catalyzed synthesis of substituted quinolones 69 by a three-component cascade annulation of nitriles, alkynes, and hypervalent iodine reagents (Scheme 92) [122]. In this case, the nitrilium intermediate XXXIX quickly reacts with acetylenes to give XL. This latter intermediate undergoes electrophilic annulation to 69. Considering the cationic mechanism of the process, the regioselectivity observed by using asymmetric alkynes in the formation of XL is favoured by electronic effects.
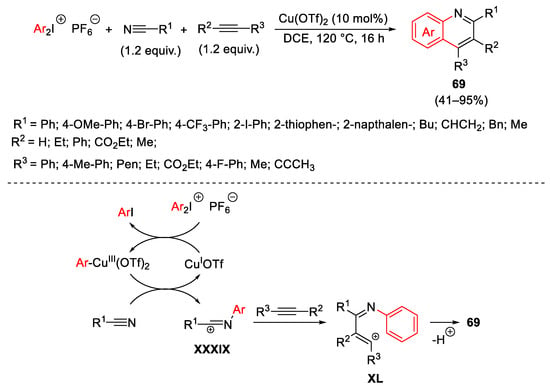
Scheme 92. Copper-catalyzed synthesis of substituted quinolines 69 by a three-component cascade annulation.
In this field, Chen et al. reported the preparation of benziodoxoles containing aliphatic amine [123] or carbazoles [124] residues to promote electrophilic amination of arenes. The reaction mechanism, involving carbazole iodane(III) reagents, is depicted in Scheme 93. Copper oxidation by the iodinane(III) reagent 70 generates the radical intermediate which in turn was trapped by the aromatic substrate. The final product was formed after the copper assisted single electron oxidation and deprotonation. This protocol could be applied to electron-rich arenes with acceptable to good yields.
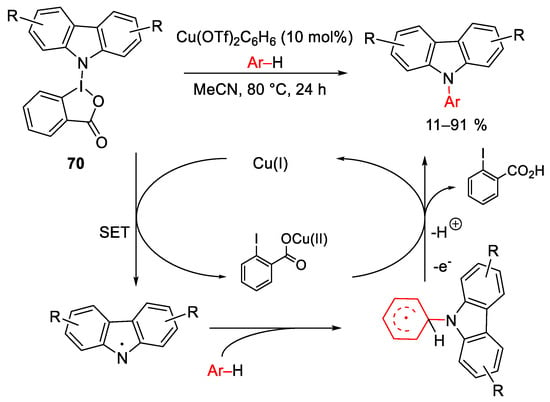
Scheme 93. Copper-catalyzed electrophilic amination of aryl compounds with carbazole-hypervalent iodane(III) reagents.
Moreover, Wen engaged cyclic diaryliodonium reagents to achieve a series of functionalized carbazoles using Cu(OAc)2 as the reaction catalyst (Scheme 94) [125]. The reaction was triggered by the reduction of the copper(II) salt. Oxidative addition of diphenyliodonium salt to Cu(I) furnished the copper(III) complex XLI which can be attacked by the nucleophile under basic conditions. The next reductive elimination of the intermediate XLII generates the aminated compound XLIII. The nitrogen metallation of XLIII, followed by intramolecular oxidative addition and reductive elimination gave access to the target carbazoles restoring the catalyst. The use of Cu/(PyBox) as co-catalytic system in the presence of a Lewis acid allows the 2,2′-amino-iodo-biaryls XLIII to be isolated with high enantioselectivity [126]. The same ring-opening reaction can be co-catalyzed by CuCl and anionic chiral cobalt(III) providing the corresponding atropisomers in high ee values [127].
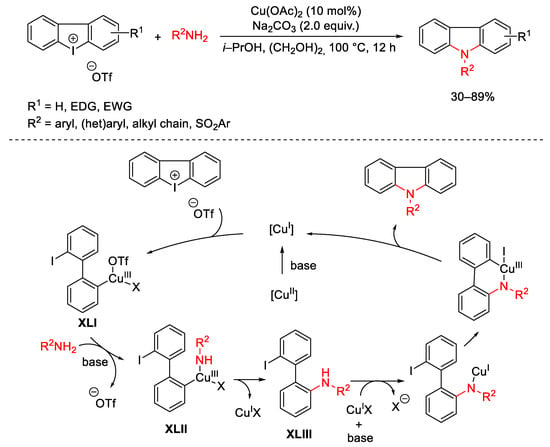
Scheme 94. One-pot copper-catalyzed amine insertion into cyclic diaryliodonium reagents.
The enantioselective Cu-catalyzed ring-opening reaction of cyclic diaryliodonium salts occurs with O-alkylhydroxylamines in the presence of 3,5-di(tert-butyl)phenyl bis-oxazoline as the optimal ligand yielding 2-hydroxyamino-2′-iodobiaryls up to 99% ee [128]. In this type of reaction, triazoles can also behave as nitrogen nucleophiles, showing excellent chemoselectivity with regard to the three different nitrogen atoms of the ring [129]. A highly chemo-and enantioselective Cu/bisoxazoline-catalyzed ring-opening of the same substrates in the presence of ketoximes of 2-(hydroxyimino)malonates was proven to be a fruitful procedure to access nitrones bearing atropisomeric biaryl moiety (Scheme 95) [130].
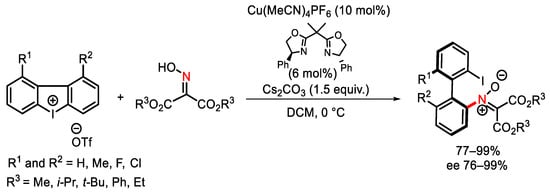
Scheme 95. Copper-catalyzed synthesis of atropisomeric nitrones.
Concerning the use of nitrogen-containing cyclic hypervalent iodane(III) reagents, Yuan developed a copper-catalyzed electrophilic N-imination of aryl(alkenyl) boronic acids with (diarylmethylene)amino-substituted benziodoxoles (Scheme 96) [131]. Experimental details suggested that the reaction could be triggered by the transmetallation of the arylboronic acid with the copper catalyst. The aryl copper(I) complex, through a single electron oxidation and radical rebounding pathway, undergoes oxidative addition with the iodane(III) reagent generating intermediate XLIV. The final reductive elimination delivered the target 71 with regeneration of the catalyst.
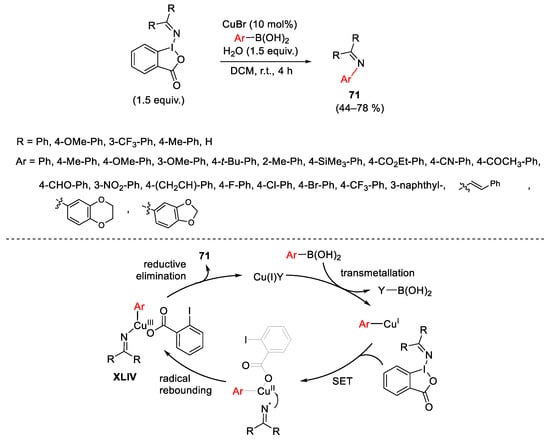
Scheme 96. Copper-catalyzed synthesis of N-aryl(alkenyl) imines using hypervalent iodane (III) reagent containing (diarylmethylene)amino groups.
Alkenyl oximes can be converted into N-aryl-isoxazolidines by initial generation of nitrone moieties through N-arylation of the oxime, which in turn undergo intramolecular 1,3-dipolar cycloaddition (Scheme 97) [132]. This is a photo-promoted reaction performed in the presence of CuBr as catalyst, diaryl iodonium salts, and bipyridine-type ligand. The reaction occurs also at the intermolecular level using N-methylmaleimide as dipolarophile.
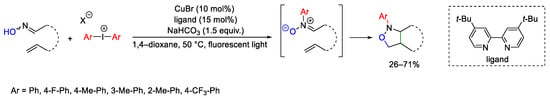
Scheme 97. Copper-catalyzed synthesis of N-aryl-sioxazolidines.
3.2. Azidation Reactions
Organic azides are of great interest in different scientific fields (i.e., chemistry, medicinal chemistry, biology, and material science). This justifies an increase in the development of new methods for their synthesis with different transition metal catalysts [133][134][135] and also with the combination of a copper catalyst with an hypervalent iodine(III) as sources of azide.
CuCl was used by Suna to promote an indirect C−H azidation of indoles using unsymmetrical indolyl(phenyl)iodonium azides 72, easily prepared by treating the corresponding indole substrates with a mixture of PhI(OAc)2 and TsOH followed by the addition of sodium azide (Scheme 98) [136]. Then, the subsequent introduction of the copper catalyst to the reaction mixture promoted fragmentation to 3-indolyl azides 73. Finally, these latter were in situ either reduced to heteroarylamines or submitted to Cu-catalyzed cycloaddition reaction to give the correspondent 1,2,3-triazole compounds. The formation of the iodonium azide 72 is consistent with products arising from SEAr reactions. On the other hand, the regioselectivity of the fragmentation of unsymmetrical iodonium salts 72 is controlled by the Cu(I) salt because in catalytic coupling reactions, electron-rich or less sterically hindered aryl moieties are selectively transferred to the metal. Furthermore, the authors indicated the π-Cu(III) complex XLV as the key reaction intermediate, which could either directly evolve to 73 or pass through the heteroarylcopper(III) XLVI due to loss of PhI followed by reductive elimination.
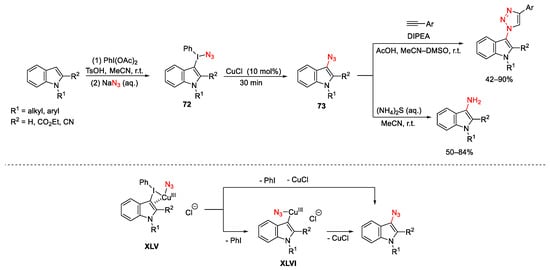
Scheme 98. Copper-catalyzed azidation of indoles.
The use of iodine(III) reagents as electrophilic source of azide allows the direct azidation of different organic compounds through a radical-based mechanism. In this field of research, the ABX has been further exploited in a range of pioneering azidation methods, considering that, in contrast to other hypervalent iodine azides, ABX is a crystalline and thermally stable solid. ABX in combination with copper(II) catalysts, has been employed to promote the direct aromatic C–H azidation of anilines in which the amino residue acts as ortho-directing group (Scheme 99) [137]. Alkoxy, halide, acetyl, and ester groups on the aryl ring are well tolerated, even though the substitution pattern of the aromatic ring was found to influence the reaction efficiency. In particular, para-substituted anilines furnish products 74 with the highest yields. Furthermore, the process runs well in the absence of any radical initiator under very mild reaction conditions, whereas complete inhibition of the reaction progress was observed in the presence of radical scavengers. Thus, the authors propose a SET process between ABX and aniline to form the aniline radical cation and the azido radical XLVII, which in turn undergoes copper-promoted O-I bond cleavage generating XLVIII. Degradation of this latter furnished the azido radical able to react with the aniline radical cation. The final deprotonation step furnishes 74.
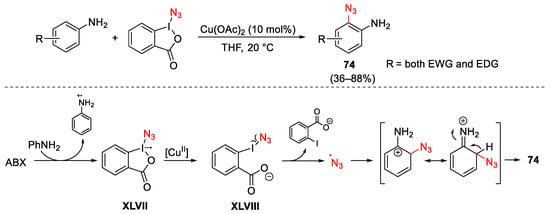
Scheme 99. Direct C–H azidation of anilines with ABX under copper catalysis.
Concerning the direct functionalization of C(sp3)-H bond, Deng and co-workers reported an enantioselective azidation of N-unprotected 3-trifluoromethyl oxindoles (Scheme 100) [138]. The reaction conditions involve Cu(CH3CN)4PF6 as catalyst, benziodazolone-based azidating reagent, and bis-oxazoline ligand XLIX.
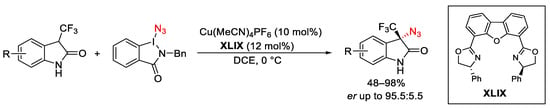
Scheme 100. Direct C–H azidation of 3-trifluoromethyl oxindoles with ABX under copper catalysis.
In the Jiao approach, Cu(acac)2 was the best copper catalyst to promote inter- and intra- molecular alkoxyazidation of indoles 75 [139]. As shown in Scheme 101 for the intramolecular version, the reported dearomatisation process occurs through copper oxidation by ABX with generation of the N3 radical. This latter was trapped by the indole ring at the nucleophilic C-3 position. After the copper-assisted single-electron oxidation, cations L and LI were produced, promoting the final intramolecular nucleophilic attack to the reaction products.
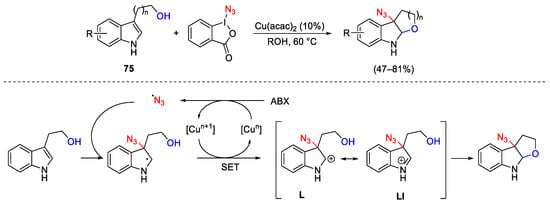
Scheme 101. Intramolecular copper-catalyzed alkoxyazidation of tryptophols.
Similarly, application of the ABX reagent to functionalization of styrene-type double bonds was explored by Greaney (Scheme 102, path a) [140]. In particular, the authors developed a copper-photoredox catalytic system using the Sauvage catalyst [Cu(dap)2Cl] and visible light to activate ABX and generate the azido radical. This latter smoothly reacted with activated alkenes in methanol to give a range of methoxy-azidated products. The same group was able to tune the scope of the photoredox catalytic system toward benzylic C-H azidation reactions by varying the reaction solvent from methanol to acetonitrile (Scheme 102, path b) [141]. The method was successfully applied to a wide range of substrates bearing different electronic properties, steric hindrance around the reactive centre, with a primary, secondary, or tertiary benzylic position. The authors suggest that the azido radical was able to extract a benzylic hydrogen atom from the starting substrate forming the benzyl radical LII. The formation of product 76 occurs through abstraction of N3 from ABX promoted by LII. Moreover, the formed iodane radical was also able to abstract a hydrogen atom from the benzylic substrate releasing LII to continue chain propagation.
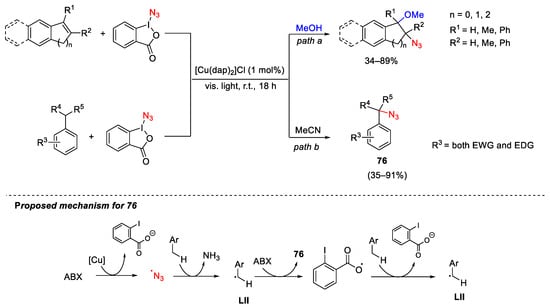
Scheme 102. Azidation of styrene-type double bond (path a) and benzylic C-H bonds (path b) with ABX under copper-photoredox catalysis.
In combination with copper(I)acetate, ABX has been employed in aminoazidation of unactivated alkenes (Scheme 103) [142]. The reported strategy shows high functional group tolerance and the scope of the process was confirmed on a range of both terminal and internal alkenes. Furthermore, besides a complete regioselectivity, very high diasteroselectivities were observed when internal alkenes were employed.
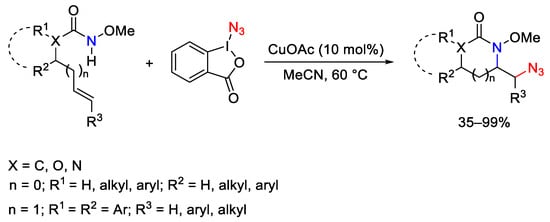
Scheme 103. Intramolecular copper-catalyzed aminoazidation of unactivated alkenes with ABX.
4. Hypervalent Iodine as Reagent in C-O Bond Formation
Moving the attention toward oxygen nucleophiles, the versatility of unsymmetrical diaryliodonium reagents was exploited again by Suma for the C-H oxyarylation of electron-rich heteroarenes or arenes (Scheme 104) [143]. In this context, the same copper-based catalytic system and the same reactivity of unsymmetrical diaryliodonium reagents previously reported in Scheme 85 for the synthesis of aryl- and heteroaryl- aniline derivatives, [104] were extended also to build diaryl or heteroaryl ether compounds. As usual, diaryliodonium salts bearing one bulky mesityl phenyl residue allow the selective transfer of the less hindered aryl ring. Furthermore, the regioselectivity of the C−H oxyarylation is controlled at the stage of the formation of iodane 77. Thus, monosubstituted phenyl compounds underwent para C−H iodination and then oxyarylation, while the C-I bond formation in poli-substituted arenes proceeded at the para-position to the strongest electron-donating group.
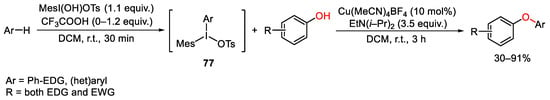
Scheme 104. Copper(I)-catalyzed C-H oxyarylation of phenol derivatives.
A combination of copper(II)-triflate catalyst together with thiophosphoramide-based co-catalyst LIII was used by Nagorny and co-workers in a process involving arylation reactions of potassium carboxylates using diaryliodonium triflates as the aryl partners (Scheme 105) [144]. The authors attributed the role of the co-catalyst LIII to the solvation of both the diaryliodonium- and organocopper- counterion (OTf−). This device resulted in milder reaction conditions and increased the scope of the reaction.
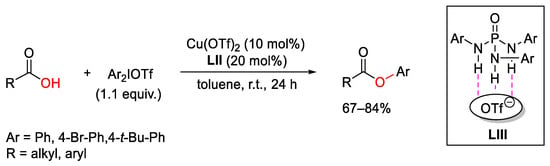
Scheme 105. Copper(II)-catalyzed arylation of carboxylates.
In 2017, Zhang and co-workers reported a copper-catalyzed diphenylation of aliphatic or (hetero)aromatic carboxylic acids with cyclic diaryliodonium reagents (Scheme 106) [145]. This one-step procedure allowed the synthesis of biphenyl esters with an iodo-substituent which could be easily transformed. Two years later, the same research group developed the enantioselective synthesis of acyloxylated 2-iodobiaryl working with Cu(OAc)2 as catalyst and with a chiral bis(oxazoline) as ligand [146].
Scheme 106. Copper(II)-catalyzed synthesis acyloxylated 2-iodobiaryl.
Copper-catalysts were also able to efficiently promote oxyarylation of alkyl phosphonates using hypervalent diaryliodonium compounds (Scheme 107) [147]. Furthermore, Feringa and Fananas-Mastral synthesised mixed alkyl aryl phosphonates 78 in a rapid, clean and easy manner via a one-step synthetic procedure using CuCl in combination with a hindered non-nucleophilic base (2,6-di-tert-butylpyridine—dtbpy). This catalytic system worked well on a wide range of phosphonates independent from the electronic properties of the substituent on the diaryl iodonium reagent employed. Experimental data suggested to the authors that the reaction pathway passed through an initial oxidative addition of the Cu(I) catalyst to the diaryliodonium salt allowing the related aryl-Cu(III) complex. This latter underwent nucleophilic attack by the phosphonate, forming intermediate LIV that, successively, evolved in LV via a base-facilitated substitution reaction of one alkyl groups by the triflate anion following a SN1-type mechanism. Reductive elimination furnished 78 restoring the Cu(I) catalyst. Compounds 78 play a pivotal role in nucleoside synthesis.
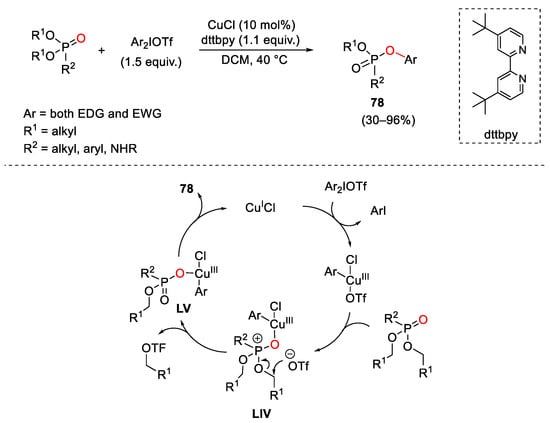
Scheme 107. Copper(I)-catalyzed C-H oxyarylation of phosphonates with diaryliodonium triflates reagents.
Based on a similar concept, a strategy to access esters of 2′-iodosubstituted biarylphosphinic acids was developed (Scheme 108) [148]. The cyclic diaryliodonium salts were opened with phosphinic acids and the best results were achieved working with CuI as catalyst in the presence of triethylamine in THF at 80 °C.
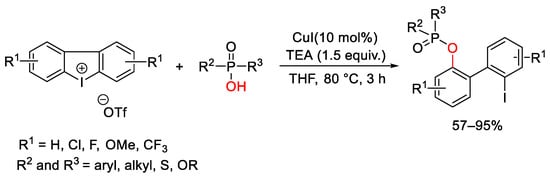
Scheme 108. CuI-catalyzed synthesis of esters of 2′-iodosubstituted biarylphosphinic acids.
An analogous Cu-catalyzed enantioselective oxidative ring-opening of cyclic diaryliodonium salts with diarylphosphine oxides allows the synthesis of atropisomeric 2′-iodo biaryl phosphine oxides, which in turn can be transformed into 2′-hydroxy biaryl phosphine oxides with total chirality retention [149].
This strategy was then extended by Fananas-Mastral to the direct alkenylation of phosphonates by using unsymmetric styryl(aryl)iodonium reagents (Scheme 109) [150]. As usual, the presence of a non-transferable mesityl ligand on the asymmetric iodonium salt was crucial for the outcome of the reaction. In this way, a series of styryl phosphonates was isolated with fair to very good yields.
Scheme 109. Copper(I)-catalyzed C-H oxyalkenylation of phosphonates with hypervalent iodine reagents.
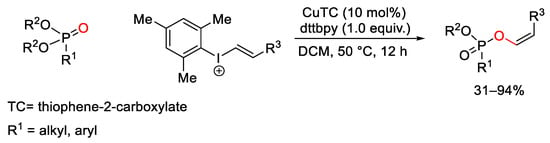
The reactivity of unsymmetrical vinyl(aryl)iodonium triflates was explored also by Gaunt for the synthesis of highly functionalized tri-, and tetra- substituted alkenyl triflates 79 [151]. In particular, copper(I) catalysts enable the electrophilic carbofunctionalization of alkynes with vinyliodonium triflates (Scheme 110). Experimental results indicated to the authors the formation of the high oxidation state Cu(III) complex LVI. This could engage the alkyne, promoting the insertion of the aryl-Cu(III) species to the carbon−carbon triple bond leading LVII. The final reductive elimination furnished 79 restoring the catalyst.
Scheme 110. Copper-catalyzed synthesis of alkenyl triflates using vinyl(aryl)iodonium triflate reagents.
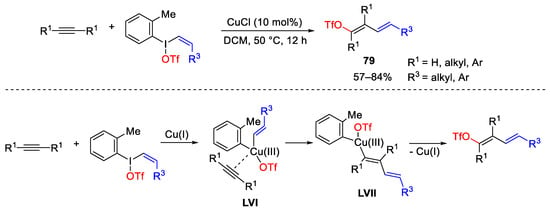
Cyclic diaryliodonium reagents could act as synthons for cascade transformations involving C-O bond formation. In particular, Zhu was able to construct biaryl oxazoles through an oxidative ring-opening/cyclization reaction of cyclic diaryliodonium compounds bearing a benzamido residue in the ortho-position to the iodine centre (Scheme 111) [152]. The reported intramolecular oxidation could be carried out in the presence of bis(oxazoline)-based chiral ligands (R,R-Ph-box) for asymmetric conversions. Most probably, the asymmetric ring-opening/cyclisation reaction involves the interaction of the Cu(I)/bisoxazoline complex with the diaryl iodium salt leading the formation of the less hindered Cu(III) complex LVIII. The catalytic system was functional-group-tolerant and allowed the isolation of differently substituted oxazole biaryls in up to 99% of both yield and enantiomeric excess. Furthermore, concomitant alkynylation of the iodine atom occurs when palladium acetate and alkyne compounds were added to the reaction mixture [153].
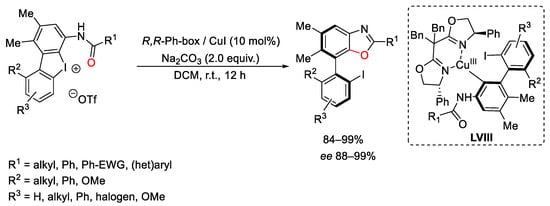
Scheme 111. Copper-catalyzed benzoxazoles formation with cyclic diaryliodonium compounds.
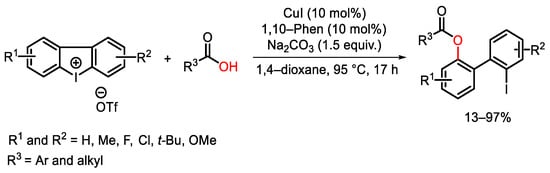
Zhu research group and other take advantages to the versatility of cyclic diaryliodonium reagents to tune a copper(II)-catalyzed tandem strategy for the synthesis of polycyclic heteroarenes from stable cyclic (het)aryl, aryl or diaryl iodoniums compounds using water as the oxygen source [154][155][156]. As reported in Scheme 112, the tricyclic (het)aryl, aryl iodonium salts 80 were easily prepared by MCPBA- promoted oxidation of the correspondent ortho-iodo heteroaryl-aryl precursor under acidic conditions. The catalytic system was generally functional-group-tolerant and allowed the isolation of complex oxygen-containing heterocycles. Furthermore, the reaction was triggered by the reduction of the copper(II) salt to the active copper(I) catalytic specie. Oxidative addition of 80 to Cu(I) furnished the copper(III) complex LIX which can be attacked by a water nucleophile. The next reductive elimination generated intermediate LX. The oxygen metalation of LX, followed by intramolecular oxidative addition and reductive elimination gave access to the target heteroarenes 81, restoring the catalyst.
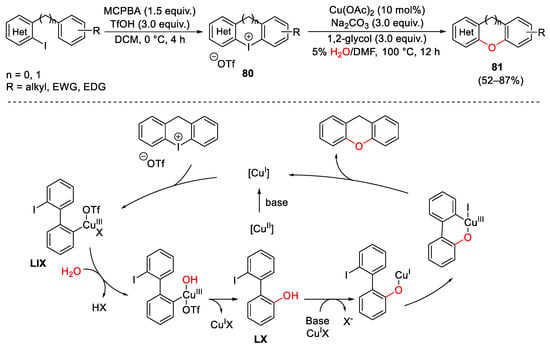
Scheme 112. Copper-catalyzed water insertion into cyclic diaryl iodonium reagents.
Alkoxy alkynes were submitted to the intramolecular etherification reaction in the presence of diaryliodonium salt [52]. In these conditions the reaction pathway afforded an oxo-heterocycle 82 involving the formation and the cleavage of a C-O bond (Scheme 113).
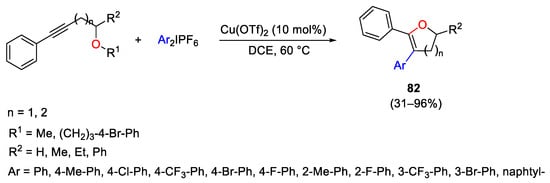
Scheme 113. Copper-catalyzed intramolecular aryl-etherification of alkoxyl alkynes.
The proposed mechanism showed the formation in situ of the Ar-Cu(III) species, easily attacked by alkynes producing vinyl-copper(III) species LXI (Scheme 114). The subsequent elimination of CuX and the intramolecular attack by the oxygen atom, followed by the C-O bond cleavage afforded the final product 83.
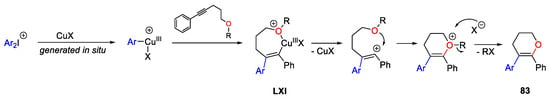
Scheme 114. Possible mechanism for the intramolecular aryl-etherification of alkoxyl alkynes.
5. Hypervalent Iodine as Reagent in C-S Bond Formation
Diaryl tricyclic iodonium salts with general formula 84 could react with various sulphur nucleophiles allowing access to a broad range of poly-heterocyclic or biaryl frameworks [157][158][159][160]. Furthermore, a remarkable Cu-catalyzed enantioselective synthesis of diarylmethanes in the presence of bis(oxazoline) chiral ligands LXII using thio-carboxylate nucleophiles was described by Gu in 2018 (Scheme 115) [161]. The authors attributed the stereochemical course of the process to steric effect associated with both the shape of 84 and the geometry of the Cu(I)/L complex. In particular, the Cu(I)/L complex underwent oxidative addition from the less sterically hindered side of 84. The next coordination of the nucleophile to the Cu(III) centre followed by reductive elimination, gave the final products.
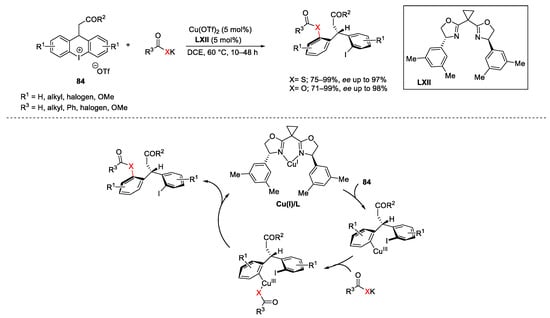
Scheme 115. Cu-catalyzed enantioselective diarylmethanes formation.
Once again—as nitrogen or oxygen nucleophiles—Cu-catalyzed reaction of diaryliodonium salts were conducted with a sulphur nucleophile (Scheme 116) [162]. In a study the authors observed that the yields of the desired trifluoromethylsulfones were independent of the nature of the diaryliodonium counter-anion. This data is in contrast to the results concerning other heteroatom-based nucleophiles.
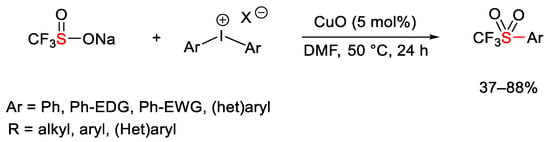
Scheme 116. Copper-catalyzed synthesis of aryltrifluoromethylsulfones.
Moving the attention toward other cyclic hypervalent iodine reagents, Waser described an efficient copper-catalyzed ring opening of sulphur heterocycles using ethynylbenziodoxolone EBX (Scheme 117) [163]. The reaction proceeds through EBX activation by the copper(I) catalyst, forming either the iodonium salt LXIII or the copper(III) complex LXIV via an oxidative transfer. In the latter case, coordination of the copper(III) ion to the sulphur heterocycle followed by reductive elimination furnished sulfonium LXV. On the other hand, formation of LXIII involved the direct formation of LXV. This underwent a ring opening by nucleophilic attack of the copper carboxylate LXVI, affording functionalized building block 85 and restoring the catalyst. The authors did not exclude a parallel radical pathway, considering that the addition of the radical scavenger TEMPO furnished the target 85 in lower yield.
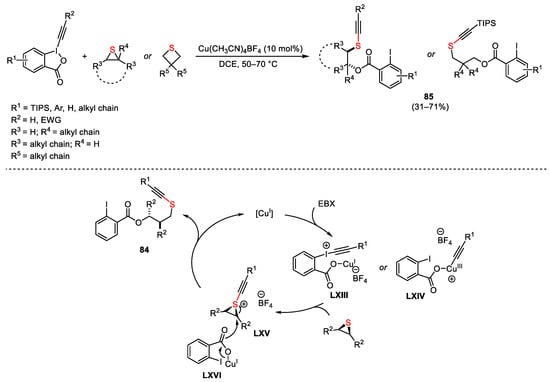
Scheme 117. Copper-catalyzed ring opening of sulphur heterocycles using EBX reagent.
A synthetic protocol for the preparation of diarylannulated sulfones through SO2/I exchange of diaryliodonium salts in the presence of Na2S2O5 was proposed by Jiang and co-workers (Scheme 118) [164]. Cu(OTf)2 as catalyst and 1,10-phenanthroline were used with Na2S2O5 as more easily handled surrogate of SO2, TBAB to increase the solubility of salts and K3PO4 as base to give products with good to excellent yields.
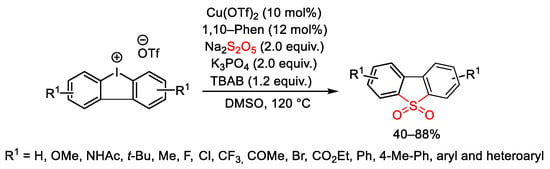
Scheme 118. Copper-catalyzed synthesis of diarylannulated sulfones through SO2/I exchange of cyclic diaryliodonium salts.
In 2013, Shibata reported an appealing electrophilic trifluoromethylthiolation of various nucleophiles with hypervalent iodonium ylide 86 in the presence of copper(I) chloride (Scheme 119) [165]. The authors envisioned that the reaction mechanism could involve the reactive trifluoromethylthio specie LXX generated from 86 by copper(I)-catalyzed carbene-mediated reduction. Furthermore, carbene LXVII was in equilibrium with thioxirene LXVIII that, in turn, was subjected to a rearrangement process generating thioperoxoate LXIX passing through the corresponding sulfoxide. In the presence of a base salt, LXX was formed and attacked by the nucleophile to afford the final products 87. When the reaction was conducted in the absence of the base, LXIX could generate the target products 87 through a single-electron transfer or an electrophilic path.
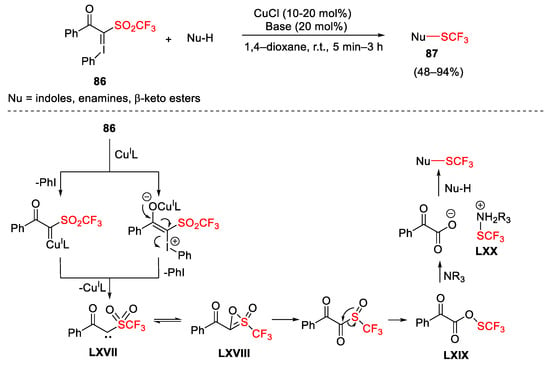
Scheme 119. Copper-catalyzed trifluoromethyl thiolation of nucleophiles with hypervalent iodonium ylide.
It should be mentioned how the well-established procedure of opening cyclic diaryliodonium salts with nucleophiles can also be achieved by employing CsSCF3 to give trifluoromethylthiolated biaryl atropisomers in high yield and enantioselectivity (Scheme 120) [166].
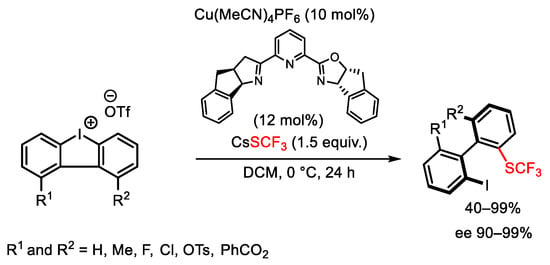
Scheme 120. Atroposelective synthesis of trifluoromethylthiolated biaryls.
To conclude the description of C-S bond construction under copper catalysis using hypervalent iodonium reagents, this approach was used for the synthesis of quinoline derivatives combining readily available alkynes, isothiocyanates, and diaryliodonium salts [167]. The reaction afforded the desired products through the formation of two C-C bonds and one C-S bond in single synthetic step (Scheme 121). The same one-pot approach was chosen for the synthesis of isocoumarins starting from the corresponding diaryliodonium salts and alkynes [168] or for the synthesis of triazolophenanthridines working in the presence of alkynes, diaryliodonium salts, and sodium azide [169].
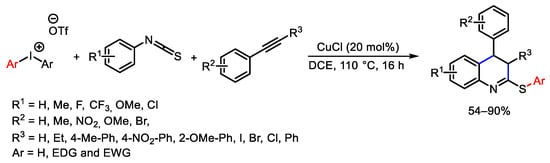
Scheme 121. Synthesis of quinoline derivatives.
6. Hypervalent Iodine as Reagent in C-X Bond Formation
The versatility of hypervalent iodine reagents was exploited for the halo-functionalization of a range of sp2 C-H bonds. However—although halogenated compounds are widely studied and synthesized with copper catalysts [170][171][172]—the use of iodines(III) in combination with copper catalysts is rather limited.
The fluoro-iodine analogue of the Togni reagent I (Scheme 122) was used for carbo-fluorination of terminal alkenes under copper catalysis by Szabò research group [173]. In particular, [Cu(MeCN)4]BF4 proved to be the best catalyst for intramolecular carbo-cyclization/fluorination of alkenyl malonate compounds.
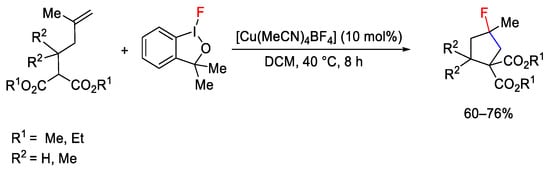
Scheme 122. Hypervalent iodine as reagent in fluorocyclization.
In the field of C-Cl bond formation, Perumgani described a direct copper(I)-mediated chlorination protocol of nitrogen-containing aromatic compounds using the chloro-iodine analogue of the Togni II reagent (Scheme 123) [174]. In this process the copper(I) catalyst promoted the homo-cleavage of the Cl-I bond in 88. Then, the generated Cu(II) and Cl radical species were coordinated by the aromatic substrate, generating the final product after copper assisted single electron oxidation and deprotonation. The reaction proceeds in good yields and complete regioselectivity. The authors suggested that the selectivity of the process could be ascribed to a pyridyl coordinated Cu-intermediate able to undergo intramolecular chlorine transfer. Ortho-chlorination occurred when 2-aryl-N-aromatic compounds were used, while remote C5 chlorination took place on quinoline substrates. The activity of the catalyst (CuI) was enhanced by the presence of K2S2O8 as an additive. Furthermore, the iodine (III) reagent is stable under the reaction conditions and could be employed in large-scale syntheses.
Scheme 123. Regioselective chlorination of C-H aromatic bonds.
As regards the C-I bond construction, an iodinative ring-opening of cyclic diaryliodonium triflates was reported by Yoshikai [175]. The reaction between aryl-substituted cyclic hypervalent iodines afforded the correspondent 2,2′-diiodobiaryl product under mild conditions in the presence of a copper/diamine catalytic system and tetra butyl ammonium iodide (TBAI) as the iodide source (Scheme 124). The oxidative addition of the catalyst to the aryl-I(III) bond generated the cupro(III) LXXI which, in turn, could be intercepted by the iodide anion. The final reductive elimination furnished the 2,2′-diiodobiaryl, regenerating the catalyst. The process could be scaled up and, at least, allow the preparation of tetraiodoteraryls which are smart precursors of heterofluorene compounds.
Scheme 124. Iodinative ring opening of cyclic hypervalent iodine reagents.
The same approach starting from cyclic diaryliodonium salts, performed in the presence of CuI with tetrabutylammonium halides and the chiral bisoxazoline ligand in hexafluoroisopropanol as solvent, was used for a more general access to axially chiral 2,2′-dihalobiaryls in good to excellent yields and excellent enantioselectivities [176].
7. Hypervalent Iodine as Reagent in C-P Bond Formation
Procedures for copper-mediated coupling of phosphorus nucleophiles with diaryliodonium reagents were also disclosed.
In 2013, Zhao reported a copper(I)chloride P-arylation process of achiral diaryl phosphine oxides and H-phosphonates nucleophile with symmetrical and unsymmetrical diaryliodonium salts (Scheme 125) [177]. The reaction proceeded at room temperature, in short reaction time and with a broad reaction scope. Furthermore, when unsymmetrical iodonium salts were used, reaction occurred preferentially on the sterically hindered—or the more electron-deficient—aromatic ring.
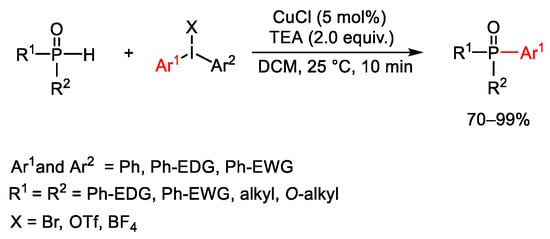
Scheme 125. Copper catalyzed P-Arylation using diaryliodonium reagents.
A related enantioselective strategy was then described by Gaunt [178]. In this process, the target products were isolated using copper(I) triflate as the catalyst in the presence of bis(oxazoline)-based chiral ligands (S,S-PhPy-box) for asymmetric conversions (Scheme 126). The catalytic system was functional-group-tolerant and allowed the isolation of differently substituted tertiary phosphine oxides in up to 98% of both yields and enantiomeric excess. Even though insight into the reaction mechanism are still in progress, most probably, the asymmetric P-arylation involved the interaction of the Cu(I)/bisoxazoline complex with the diaryliodium salt forming the less hindered Cu(III) species LXXII. This latter, in turn, could bind to the starting phosphine oxides. The stereo controlled reductive elimination generated the target products.
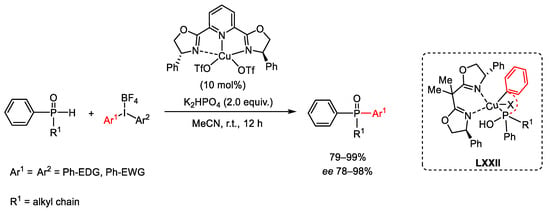
Scheme 126. Enantioselective copper-catalyzed P-Arylation using diaryliodonium reagents.
8. Hypervalent Iodines as Oxidant
The use of hypervalent iodine as oxidant to provide coupling reactions involving the formation of different C-C and C-N bonds is an elegant and efficient synthetic pathway to afford heterocyclic systems.
In this context, the use of N-sulfonyl 2-amidobiphenyls 89 as substrates allowed carbazoles 90 to be obtained, under both Cu-catalyzed and metal-free conditions, with generally lower yields in the latter case [179]. The observed kinetic insensitivity to the copper catalyst led to propose that the copper species worked as a Lewis acid to activate hypervalent iodine(III) oxidant. Moreover, when the radical scavenger was added to the reaction mixture, the reaction was significantly decreased, suggesting that a radical intermediate was involved. The reaction realized in the presence of Cu(OTf)2 in DCE at 50 °C was compatible with electron-donating substituents at the nucleophilic phenyl part (right side) and the electron-withdrawing groups on the “left” rings facilitated the cyclization (Scheme 127).
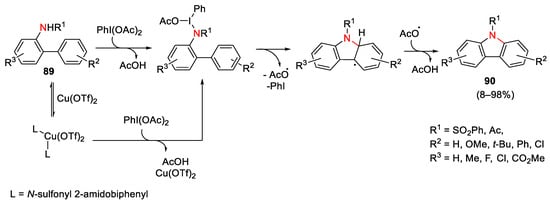
Scheme 127. Mechanism for the synthesis of carbazole under Cu-catalyzed or metal-free conditions using hypervalent iodine (III) as oxidant.
Another polyheterocyclic system such as the substituted benzo[4,5]imidazo[1,2-c]quinazoline was prepared starting from 4-anilinoquinazoline, exploiting as a key step the oxidative coupling catalyzed by Cu(OTf)2 and hypervalent iodine oxidant to form a C-N bond [180] (Scheme 128).
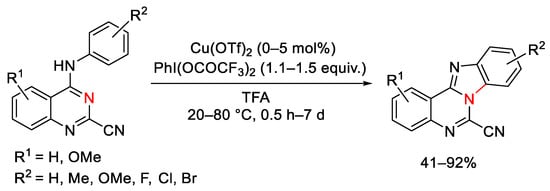
Scheme 128. Copper(II)-mediated oxidative ring closure of 4-anilinoquinazoline-2-carbonitriles.
Radical decarboxylative process involving C-N bond formation has been studied and applied at the synthesis of pyrrolidines and piperidines [181][182]. Different complex mechanisms could be hypothesized where the oxidant PhIO and the copper serve to obtain decarboxylation followed by C-N coupling, also considering the essential presence of the picolinamide (PA) as directing group for the site-selectivity (Scheme 129).
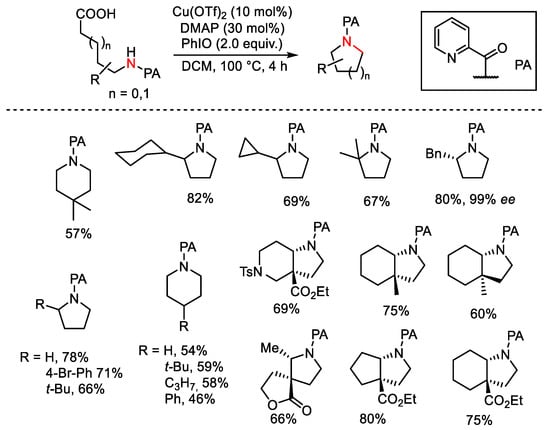
Scheme 129. Copper-catalyzed intramolecular decarboxylative C-N coupling of aliphatic carboxylic acids.
Substituted pyrrolidine-2,4-diones were obtained also by intramolecular amidation of β-dimethylamino-propenoyl alkylamides 91, in the presence of PIFA and TFA as additive, under CuBr2 catalysis [183]. The reaction proceeds in good yields under mild reaction conditions, through a radical mechanism involving the dihydrofuran-3-one LXXIII formation followed by ring opening in the presence of copper halide to give a radical LXXIV, the cyclization of which afforded LXXV. The pyrrolidine-2,4-dione 92 was obtained by final hydrolysis. The presence of substituents in the α-position increases the efficiency of the cyclization (Scheme 130).
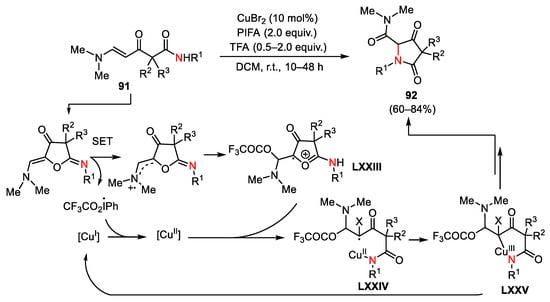
Scheme 130. Plausible mechanism for the formation of pyrrolidine-2,4-diones.
Enantioselective cyclopropanation of alkenes has been developed using an iodonium ylides derived from methyl nitroacetate with a chiral copper complex, obtaining trans nitrocyclopropane carboxylates 93 in high diastereo- and enantioselectivity [184]. The reaction involved the use of CuCl and AgSbF6 to generate the CuSbF6 catalyst, that gave the best results in term of enantioselectivity, bis(oxazoline) ligand LXXVI, PhIO, and Na2CO3 as base in benzene as solvent (Scheme 131).
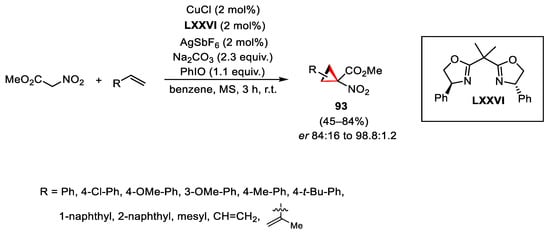
Scheme 131. Copper-catalyzed cyclopropanation reaction.
Cyclopropanation was also obtained starting from an alkene, Wittig reagent and iodosotoluene as oxidant, in the presence of Cu(tfacac)2 catalyst [185]. The stereochemistry of the alkenes gave, in general, a 2/3:1 mixture of diastereomers (Scheme 132). The authors hypothesized, from the Wittig precursor and the oxidant, the formation of a transient monocarbonyl iodonium ylide LXXVII which can generate a metallocarbene LXXVIII through the reaction with the copper catalyst and decomposition. The carbene then afforded cyclopropanation by reaction with the unsaturated system, regenerating the catalyst.
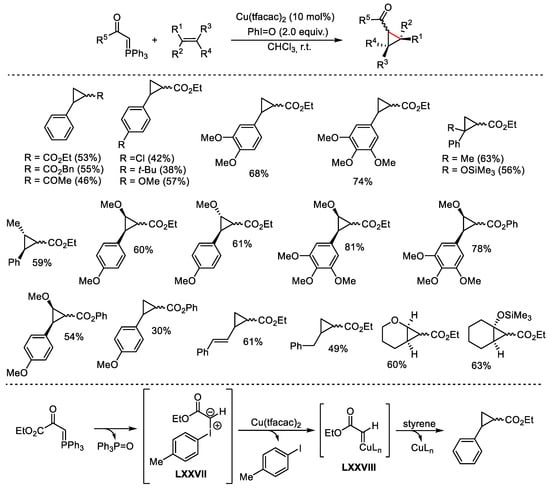
Scheme 132. Cyclopropanation of alkenes in the presence of catalytic Cu(tfacac)2 and iodosotoluene.
A particular synthesis of α-keto amides was realized starting from enaminones by means of oxidation in the presence of hypervalent iodine and copper catalysis [186]. The presence of water was mandatory, acting as the oxygen source during the generation of the carbonyl group after the cleavage of C-C double bond (Scheme 133).
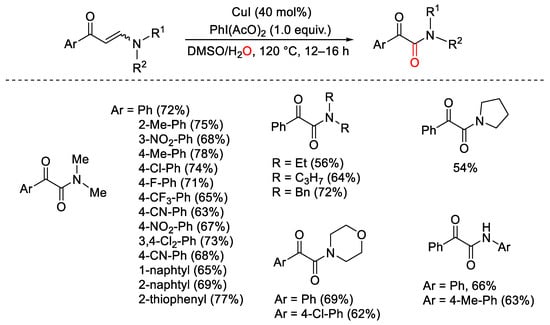
Scheme 133. Synthesis of α-keto amides from enaminones under the copper/PhI(OAc)2 catalytic system.
A particular reactivity of 2-benzylamino-phenols 94 mediated by PIDA was reported by a research group, resulting in the synthesis of oxazolo-phenoxazines 95 through a cascade three-step C-H functionalizations [187]. The process involves the dimerization of the substrate after an initial oxidation step, whereas the formation of the terminal five-membered ring is due to the action of the copper catalysis (Scheme 134).
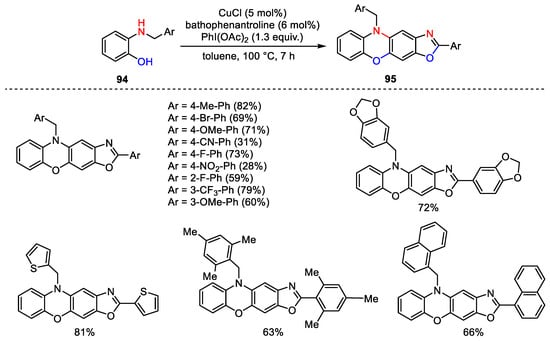
Scheme 134. Dimerization/cyclization of 2-benzylaminophenols.
The reaction performed under standard conditions on the N,N′-dibenzyl-1,2-benzenediamines 96, although gave rise to a dimerization process, did not provide tetracyclic fused-ring system but led only to the formation of benzo[d]imidazol-6-yl)benzene-1,2-diamine products 97 (Scheme 135). Probably, the presence of a second benzylamino substituent instead of the hydroxyl group inhibited the formation of the second C-N bond, hampering access to the phenazine nucleus.
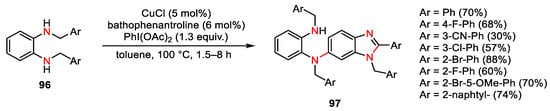
Scheme 135. Dimerization/cyclization of N,N′-dibenzyl-1,2-benzenediamines.
The C-O bond formation was reported in the alkoxylation of S,S-functionalized internal olefins [188]. In this case, the Cu(OH)2 was used as catalyst and besides the (diacetoxyiodo)benzene as oxidant, also benzoquinone (BQ) was used as co-oxidant (Scheme 136). The present procedure also provided a route to alkoxylated N-heterocycles.
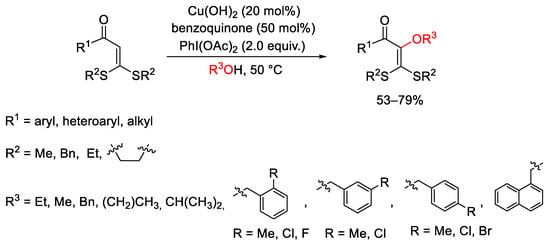
Scheme 136. Alkoxylation of S,S-functionalized internal olefins.
References
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Chichester, UK, 2013.
- Richardson, R.D.; Wirth, T. Hypervalent Iodine Goes Catalytic. Angew. Chem. Int. Ed. 2006, 45, 4402–4404.
- Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665.
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin/Heidelberg, Germany, 2003.
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1996.
- Le Du, E.; Waser, J. Recent progress in alkynylation with hypervalent iodine reagents. Chem. Commun. 2023, 59, 1589–1604.
- Simonet-Davin, R.; Waser, J. Azidation with Hypervalent Iodine Reagents. Synthesis 2023, 55, 1652–1661.
- Yoshimura, A.; Zhdanlin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435.
- Shetgaonkar, S.E.; Singh, F.V. Catalytic stereoselective synthesis involving hypervalent iodine-based chiral auxiliaries. Org. Biomol. Chem. 2023, 21, 4163–4180.
- Singh, F.V.; Shetgaonkar, S.E. Progress in organocatalysis with hypervalent iodine cataysts. Chem. Soc. Rev. 2022, 51, 8102–8139.
- Rani, N.; Soni, R.; Sihag, M.; Kinger, M.; Aneja, D.K. Combined Approach of Hypervalent Iodine Reagents and Transition Metals in Organic Reactions. Adv. Synth. Catal. 2022, 364, 1798–1848.
- Shetgaonkar, S.E.; Mamgain, R.; Kikushima, K.; Dohi, T.; Singh, F.V. Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents. Molecules 2022, 27, 3900.
- Sun, T.-Y.; Wang, X.; Geng, H.; Xie, Y.; Wu, Y.-D.; Zhang, X.; Schaefer, H.F. III Why does Togni’s Reagent I exist in the High-Energy Hypervalent Iodine Form? Re-Evaluation of Benziodoxole Based Hypervalent Iodine Reagents. Chem. Commun. 2016, 52, 5371–5374.
- Zhdankin, V.V. Benziodoxole-Based Hypervalent Iodine Reagents in Organic Synthesis. Curr. Org. Synth. 2005, 2, 121–145.
- Nicolaou, K.C.; Mathison, C.J.N.; Montagnon, T. o-Iodoxybenzoic Acid (IBX) as a Viable Reagent in the Manipulation of Ni-trogen- and Sulfur-Containing Substrates: Scope, Generality, and Mechanism of IBX-Mediated Amine Oxidations and Dithiane Deprotections. J. Am. Chem. Soc. 2004, 126, 5192–5201.
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. Iodine(V) Reagents in Organic Synthesis. Part 3. New Routes to Heterocyclic Compounds via o-Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244.
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358.
- Meyer, S.D.; Schreiber, S.L. Accelerationof the Dess-Martin oxidation by water. J. Org. Chem. 1994, 59, 7549–7552.
- Duschek, A.; Kirh, S.F. 2-Iodoxybenzoic Acid—A Simple Oxidant with a Dazzling Array of Potential Applications. Angew. Chem. Int. Ed. 2011, 50, 1524–1552.
- Wirth, T. IBX—New Reactions with an Old Reagent. Angew. Chem. Int. Ed. 2001, 40, 2812–2814.
- Ochiai, M.; Sueda, T.; Miyamoto, K.; Kriprof, P.; Zhdankin, V.V. Trans Influences on Hypervalent Bonding of Aryl λ3-Iodanes: Their Stabilities and Isodesmic Reactions of Benziodoxolones and Benziodazolones. Angew. Chem. Int. Ed. 2006, 45, 8203–8206.
- Zhdankin, V.V.; Kuehl, C.J.; Krasunsky, A.P.; Bolz, J.T.; Simonsen, A.J. 1-(Organosulfonyloxy)-3(1H)-1,2-benziodoxoles: Preparation and Reactions with Alkynyltrimethylsilanes. J. Org. Chem. 1996, 61, 6547–6551.
- Ochiai, M.; Masaki, Y.; Shiro, M. Synthesis and Structure of 1-Alkynyl-1,2-Benziodoxol-3(1H)-Ones. J. Org. Chem. 1991, 56, 5511–5513.
- Zhdankin, V.V.; Krasutsky, A.P.; Kuehl, C.J.; Simonsen, A.J.; Woodward, J.K.; Mismash, B.; Bolz, J.T. Preparation, X-Ray Crystal Structure, and Chemistry of Stable Azidoiodinanes Derivatives of Benziodoxole. J. Am. Chem. Soc. 1996, 118, 5192–5197.
- Zhdankin, V.V.; Kuehl, C.J.; Krasutsky, A.P.; Bolz, J.T.; Mismash, B.; Woodward, J.K.; Simonsen, A.J. 1-Cyano-3-(1H)-1,2-Benziodoxols: Stable Cyanoiodinanes and Efficient Reagents for Direct N-Alkyl Cyanation of N,N-Dialkylarylamines. Tetrahedron Lett. 1995, 36, 7975–7978.
- Daugulis, O.; Do, H.-Q.; Shabashov, D. Palladium- and Copper-Catalyzed Arylation of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2009, 42, 1074–1086.
- Fañanás-Mastral, M. Copper-Catalyzed Arylation with Diaryliodonium Salts. Synthesis 2017, 49, 1905–1930.
- Loro, C.; Oble, J.; Foschi, F.; Papis, M.; Beccalli, E.M.; Giofrè, S.; Poli, G.; Broggini, G. Acid-mediated decarboxylatiove C-H coupling between arenes and O-allyl carbamates. Org. Chem. Front. 2022, 9, 1711–1718.
- Phipps, R.J.; Gaunt, M.J. A Meta-Selective Copper-Catalyzed C-H Bond Arylation. Science 2009, 323, 1593–1597.
- Chen, B.; Hou, X.-L.; Li, Y.-X.; Wu, Y.-D. Mechanistic Understanding of the Unespected Meta Selectivity in Copper-Catalyzed Anilide C-H Bond Arylation. J. Am. Chem. Soc. 2011, 133, 7668–7671.
- Duong, H.A.; Gilligan, R.E.; Cooke, M.L.; Phipps, R.J.; Gaunt, M.J. Copper(II)-Catalyzed meta-Selective Direct Arylation of α-Aryl Carbonyl Compounds. Angew. Chem. Int. Ed. 2011, 50, 463–466.
- Ciana, C.-L.; Phipps, R.J.; Brandt, J.R.; Meyer, F.-M.; Gaunt, M.J. A Highly Para-Selective Copper(II)-Catalyzed Direct Arylation of Aniline and Phenol Derivatives. Angew. Chem. Int. Ed. 2011, 50, 458–462.
- Phipps, R.J.; Grimster, R.P.; Gaunt, M.J. Cu(II)-Catalyzed Direct and Site-Selective Arylation of Indoles Under Mild Conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174.
- Liang, H.; Zhu, G.; Pu, X.; Qiu, L. Copper-Catalyzed Enantioselective C-H Arylation between 2-Arylindoles and Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9246–9250.
- Pitts, A.K.; O’Hara, F.; Snell, R.H.; Gaunt, M.J. A Concise and Scalable Strategy for the Total Synthesis of Dictyodendrin B Based on Sequential C-H- Functionalization. Angew. Chem. Int. Ed. 2015, 54, 5451–5455.
- Yang, Y.; Gao, P.; Zhao, Y.; Shi, Z. Regiocontrolled Direct C-H Arylation of Indoles at the C4 and C5 Positions. Angew. Chem. Int. Ed. 2017, 56, 3966–3971.
- Yang, Y.; Li, R.; Zhao, Y.; Zhao, D.; Shi, Z. Cu-Catalyzed Direct C6-Arylation of Indoles. J. Am. Chem. Soc. 2016, 138, 8734–8737.
- Modha, S.G.; Greaney, M.F. Atom-Economical Transformation od Diaryliodoniu, Salts: Tandem C-H and N-H Arylation of Indoles. J. Am. Chem. Soc. 2015, 137, 1416–1419.
- Allen, A.E.; MacMillan, D.W.C. Enantioselective α-Arylation of Aldehydes via the Productive Merger of Iodonium Salts and Organocatalysis. J. Am. Chem. Soc. 2011, 133, 4260–4263.
- Pan, J.-L.; Chen, T.; Zhang, Z.-Q.; Li, Y.-F.; Zhang, X.-M.; Zhang, F.-M. A Cu-mediated one-pot Michael addition/α-arylation strategy using a diaryliodonium salt: A direct and efficient approach to α-aryl-β-substituted cyclic ketone scaffolds. Chem. Commun. 2016, 52, 2382–2385.
- Bigot, A.; Williamson, A.E.; Gaunt, M.J. Enantioselective α-Arylation of N-Acyloxazolidinones with Copper(II)-bisoxazoline Catalysts and Diaryliodonium Salts. J. Am. Chem. Soc. 2011, 133, 13778–13781.
- Harvey, J.S.; Simonovich, S.P.; Jamison, C.R.; MacMillan, D.W.C. Enantioselective α-Arylation od Carbonyls via Cu(I)-Bisoxazoline Catalysis. J. Am. Chem. Soc. 2011, 133, 13782–13785.
- Zhu, S.; MacMillan, D.W.C. Enantioselective Copper-Catalyzed Construction of Aryl Pyrrolindolines via an Aryla-tion-Cyclization Cascade. J. Am. Chem. Soc. 2012, 134, 10815–10818.
- Kieffer, M.E.; Chuang, K.V.; Reisman, S.E. A copper-catalyzed arylation of tryptamines for the direct synthesis of aryl pyr-roloindolines. Chem. Sci. 2012, 3, 3170–3174.
- Kieffer, M.E.; Chuangm, K.V.; Reinsman, S.E. Copper-Catalyzed Diasteroselective Arylation of Tryptophan Derivatives: Total Synthesis of (+)-Naseseazines A and B. J. Am. Chem. Soc. 2013, 135, 5557–5560.
- Phipps, R.J.; McMurray, L.; Ritter, S.; Doung, H.A.; Gaunt, M.J. Copper-Catalyzed Alkene Arylation with Diaryliodonium Salts. J. Am. Chem. Soc. 2012, 134, 10773–10776.
- Almasalma, A.A.; Mejia, E. 1-Phenyl-1,2-benziodoxol-3-(1H)-one as Synthon for Phthalide Synthesis through Pd-Free, Base-Free, Sonoganshira-Type Couple Cyclization Reaction. Eur. J. Org. Chem. 2018, 2018, 188–195.
- Xu, Z.-F.; Cai, C.-X.; Liu, J.-T. Copper-Catalyzed Regioselective Reaction of Internal Alkenes and Diaryliodonium Salts. Org. Lett. 2013, 15, 2096–2099.
- Walkinshaw, A.J.; Xu, W.; Suero, M.G.; Gaunt, M.J. Copper-Catalyzed Carboarylation of Alkynes via Vinyl Cations. J. Am. Chem. Soc. 2013, 135, 12532–12535.
- Peng, J.; Chen, C.; Chen, J.; Su, X.; Xi, C.; Chen, H. Cu-Catalyzed Arylcarbocyclization of Alkynes with Diaryliodonium Salts through C-C Bond Formation on Inert C(sp3)-H Bond. Org. Lett. 2014, 16, 3776–3779.
- Zhang, F.; Das, S.; Walkingshaw, A.J.; Casitas, A.; Taylor, M.; Suero, M.G.; Gaunt, M.J. Cu-Catalyzed Cascades to Carbocyles: Union of Diaryliodonium Salts with Alkenes or Alkynes Exploiting Remote Carbocations. J. Am. Chem. Soc. 2014, 136, 8851–8854.
- Chen, J.; Chen, C.; Chen, J.; Wang, G.; Qu, H. Cu-catalyzed intramolecular aryl-etherification reactions of alkoxyl alkynes with diaryliodonium salts via cleavage of a stable C–O bond. Chem. Commun. 2015, 51, 1356–1359.
- Sinai, A.; Vangel, D.; Gati, T.; Bombincz, P.; Novak, Z. Utilization of Copper-Catalyzed Carboarylation-Ring Closure for the Synthesis of New Oxazoline Derivatives. Org. Lett. 2015, 17, 4136–4139.
- Sinai, A.; Meszaros, A.; Gati, T.; Kudar, V.; Pallo, A.; Novak, Z. Copper-Catalyzed Oxidative Ring Closure and Carboarylation of 2-Ethynylanilides. Org. Lett. 2013, 15, 5654–5657.
- Collins, B.S.L.; Suero, M.G.; Gaunt, M.J. Copper-Catalyzed Arylative Meyer-Schuster Rearrangement of Propargylic Alcohols to Complex Enones Using Diaryliodonium Salts. Angew. Chem. Int. Ed. 2013, 52, 5799–5802.
- Cahard, E.; Bremeyer, N.; Gaunt, M.J. Copper-Catalyzed Intramolecular Electrophilic Carbofunctionalization of Allylic Amides. Angew. Chem. Int. Ed. 2013, 52, 9284–9288.
- Cahard, E.; Male, H.P.J.; Tissot, M.; Gaunt, M.J. Enantioselective and Regiodivergent Copper-Catalyzed Electrophilic Arylation of Allylic Amides with Diaryliodonium Salts. J. Am. Chem. Soc. 2015, 137, 7986–7989.
- Gigant, N.; Chausset-Boissarie, L.; Belhomme, M.-C.; Poisson, T.; Pannecoucke, X.; Gillaizeau, I. Copper-Catalyzed Direct Ar-ylation of Cyclic Enamides Using Diaryliodonium Salts. Org. Lett. 2013, 15, 278–281.
- Prakash, M.; Muthusamy, S.; Kesavan, V. Copper(I) Bromide Catalyzed Arylation of Cyclic Enamides and Naph-thyl-1-acetamides Using Diaryliodonium Salts. J. Org. Chem. 2014, 79, 7836–7843.
- Kumar, D.; Pilania, M.; Arun, V.; Pooniya, S. C-H arylation of azaheterocycles: A direct ligand-free and Cu-catalyzed approach using diaryliodonium salts. Org. Biomol. Chem. 2014, 12, 6340–6344.
- Hari, D.P.; Waser, J. Copper-Catalyzed Oxy-Alkynylation of Diazo Compounds with Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 2190–2193.
- Hari, D.P.; Schouwey, L.; Baber, V.; Scopelliti, R.; Fadaei-Tirani, F.; Waser, J. Ethynylbenziodazolones (EBZ) as Electrophilic Alkynylation Reagents for the Highly Enantioselective Copper-Catalyzed Oxyalkynylation of Diazo Compounds. Chem. Eur. J. 2019, 25, 9522–9528.
- Pisella, G.; Gagnebin, A.; Waser, J. Three-Component Reaction for the Synthesis of Highly Functionalized Propargyl Ethers. Chem. Eur. J. 2020, 26, 10199–10204.
- Shen, K.; Wang, Q. Copper-catalyzed aminoalkynylation of alkenes with hypervalent iodine reagents. Chem. Sci. 2017, 8, 8265–8270.
- Teodoro, B.V.M.; Silva, L.F.J. Sequential Michael Addition/Electrophilic Alkynylation: Synthesis of α-Alkynyl-β-Substituted Ketones and Chromanones. J. Org. Chem. 2018, 83, 13604–13611.
- Sun, X.; Guo, X.-Q.; Chen, L.-M.; Kang, T.-R. Synthesis, Characterization of Spirocyclic λ3-Iodanes and Their Application to Prepare 4,1-Benzoxazepine-2,5-diones and 1,3-Diynes. Chem. Eur. J. 2021, 27, 4312–4316.
- Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. Copper-Catalyzed C(sp3)-C(sp3) Bond Formation Using a Hypervalent Iodine Reagent: An Efficient Allylic Trifluoromethylation. J. Am. Chem. Soc. 2011, 133, 16410–16413.
- Parsons, A.T.; Buchwald, S.L. Copper-Catalyzed Trifluoromethylation of Unactivated Olefins. Angew. Chem. Int. Ed. 2011, 50, 9120–9123.
- Lonca, G.H.; Ong, D.Y.; Tran, T.M.H.; Tejo, C.; Chiba, S.; Gagosz, F. Anti-Markovinkov Hydrofunctionalization of Alkenes: Usa of a Benzyl Group as a Traceless Redox-Active Hydrogen Donor. Angew. Chem. Int. Ed. 2017, 56, 11440–11444.
- Shimizu, R.; Egami, H.; Hamashima, Y.; Sodeoka, M. Copper-Catalyzed Trifluoromethylation of Allylsilanes. Angew. Chem. Int. Ed. 2012, 51, 4577–4580.
- Wang, X.-P.; Lin, J.-H.; Zhang, C.-P.; Xiao, J.-C.; Zheng, X. Copper-catalyzed trifluoromethylation of alkenes with an electrophilic trifluoromethylating reagent. Beilstein J. Org. Chem. 2013, 9, 2635–2640.
- Weng, Z.; Li, H.; He, W.; Yao, L.-F.; Tan, J.; Chen, J.; Yuan, Y.; Huan, K.-W. Mild copper-catalyzed trifluoromethylation of terminal alkynes using an electrophilic trifluoromethylating reagent. Tetrahedron 2012, 68, 2527–2531.
- Feng, C.; Loh, T.-P. Copper-catalyzed olefinic trifluoromethylation of enamides at room temperature. Chem. Sci. 2012, 3, 3458–3462.
- Feng, C.; Loh, T.-P. Directing-Group-Assisted Copper-Catalyzed Olefinic Trifluoromethylation of Electron-Deficient Alkenes. Angew. Chem. Int. Ed. 2013, 52, 12414–12417.
- Fang, Z.; Ning, Y.; Mi, P.; Liao, P.; Bi, X. Catalytic C-H α-Trifluoromethylation of α,β-Unsaturated Carbonyl Compounds. Org. Lett. 2014, 16, 1522–1525.
- Wang, X.; Ye, Y.; Ji, G.; Xu, Y.; Zhang, S.; Feng, J.; Zhang, Y.; Wang, J. Copper-Catalyzed Direct C-H Trifluoromethylation of Quinones. Org. Lett. 2013, 15, 3730–3733.
- Deng, Q.-H.; Wadepohl, H.; Gade, L.H. Highly Enantioselective Copper-Catalyzed Electrophilic Trifluoromethylation of b-Ketoesters. J. Am. Chem. Soc. 2012, 134, 10769–10772.
- Allen, A.E.; MacMillan, D.W.C. The Productive Merger of Iodonium Salts and Organocatalysis: A Non-photolytic Approach to the Enantioselective α-Trifluoromethylation of Aldehydes. J. Am. Chem. Soc. 2010, 132, 4986–4987.
- Pair, E.; Monteiro, N.; Bouyssi, D.; Baudoin, O. Copper-Catalyzed Trifluoromethylation of N,N-Dialkylhydrazones. Angew. Chem. Int. Ed. 2013, 52, 5346–5349.
- Prieto, A.; Jearmet, E.; Monteiro, N.; Bouyssi, D.; Baudoin, O. Copper-Catalyzed b-Trifluoromethylation of Conjugated Hydrazones. Org. Lett. 2014, 16, 4770–4773.
- Prieto, A.; Bouyssi, D.; Monteiro, N. Copper-Catalyzed Trifluoromethylation od Hydrazones Leading to the Formation of Quaternary α-Trifluoromethyl Diazenes. Asian J. Org. Chem. 2016, 5, 742–745.
- Wu, S.; Li, J.; He, R.; Jia, K.; Chen, Y. Terminal Trifluoromethylation of Ketones via Selective C-C Cleavage of Cycloalkanols Enabled by Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9204–9209.
- Xiong, Y.-P.; Wu, M.-Y.; Zhang, X.-Y.; Ma, C.-L.; Huang, L.; Zhao, L.-J.; Tan, B.; Liu, X.-Y. Direct Access to α-Trifluoromethyl Enones via Efficient Copper-Catalyzed Trifluoromethylation of Meyer-Schuster Rearrangement. Org. Lett. 2014, 16, 1000–1003.
- Cai, S.; Chen, C.; Sun, Z.; Xi, C. CuCl-catalyzed ortho trifluoromethylation of arenes and heteroarenes with a pivalamido directing group. Chem. Commun. 2013, 49, 4552–4554.
- Liu, T.; Shen, Q. Copper-Catalyzed Trifluoromethylation of Aryl and Vinyl Boronic Acids with An Electrophilic Trifluoro-methylating Reagent. Org. Lett. 2011, 13, 2342–2345.
- Huang, Y.; Fang, X.; Lin, X.; Li, H.; He, W.; Huang, K.-W.; Yuan, Y.; Weng, Z. Room-temperature base-free copper-catalyzed trifluoromethylation of organotrifluoroborates to trifluoromethylarenes. Tetrahedron 2012, 68, 9949–9953.
- Zheng, H.; Huang, Y.; Wang, Z.; Li, H.; Huang, K.-W.; Yuan, Y.; Weng, Z. Synthesis of trifluoromethylated acetylenes via copper-catalyzed trifluoromethylation of alkynyltrifluoroborates. Tetrahedron Lett. 2012, 53, 6646–6649.
- Shimizu, R.; Egami, H.; Nagi, T.; Chae, J.; Hamashima, Y.; Sodeoka, M. Direct C2-trifluoromethylation of indole derivatives catalyzed by copper acetate. Tetrahedron Lett. 2010, 51, 5947–5949.
- Janson, P.G.; Ghoneim, I.; Ilchenko, N.O.; Szabó, K.J. Electrophilic Trifluoromethylation by Copper-Catalyzed Addition of CF3-Transfer Reagents to Alkenes and Alkynes. Org. Lett. 2012, 14, 2882–2885.
- Egami, H.; Shimizu, R.; Sodeoka, M. Oxytrifluoromethylation of multiple bonds using copper catalyst under mild conditions. Tetrahedron Lett. 2012, 53, 5503–5506.
- Ramkumar, N.; Baumane, L.; Zacs, D.; Veliks, J. Merging Copper(I) Photoredox Catalysis and Iodine(III) Chemistry for the Oxy-monofluoromethylation of Alkenes. Angew. Chem. Int. Ed. 2023, 62, e202219027.
- Wang, F.; Qi, X.; Liang, Z.; Chen, P.; Liu, G. Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides. Angew. Chem. Int. Ed. 2014, 53, 1881–1886.
- Ilchenko, N.O.; Janson, P.G.; Szabo, K.J. Copper-Mediated Cyanotrifluoromethylation of Styrenes Using the Togni Reagent. J. Org. Chem. 2013, 78, 11087–11091.
- Egami, H.; Kawamura, S.; Miyazaki, A.; Sodeaoka, M. Trifluoromethylation Reactions for the Synthesis of β-Trifluoromethylamines. Angew. Chem. Int. Ed. 2013, 125, 7995–7998.
- Kawamura, S.; Egami, H.; Sodeoka, M. Aminotrifluoromethylation of Olefins via Cyclic Amine Formation: Mechanistic Study and Application to Synthesis of Trifluoromethylated Pyrrolidines. J. Am. Chem. Soc. 2015, 137, 4865–4873.
- Shen, K.; Wang, Q. Copper-catalyzed aminotrifluoromethylayion of alkenes: A facile synthesis of CF3-containing lactams. Org. Chem. Front. 2016, 3, 222–226.
- Zhu, R.; Buchwald, S.L. Copper-Catalyzed Oxytrifluoromethylation of Unactivated Alkenes. J. Am. Chem. Soc. 2012, 134, 12462–12465.
- Zhu, R.; Buchwald, S.L. Enantioselective Functionalization of Radical Intermediates in Redox Catalysis: Copper-Catalyzed Asymmetric Oxytrifluoromethylation of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 12655–12658.
- Lu, D.-F.; Zhu, C.-L.; Xu, H. Copper(I)-catalyzed diastereoselective hydroxytrifluoromethylation of dienes accelerated by phosphine ligands. Chem. Sci. 2013, 4, 2478–2482.
- Yu, Q.; Ma, S. Copper-Catalyzed Cyclic Oxytrifluoromethylation of 2,3-Allenoic Acids to Trifluoromethylated Butenolides. Chem. Eur. J. 2013, 19, 13304–13308.
- He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Wang, Y.-Q.; Luo, J.-Y.; Liu, X.-Y.; Liang, Y.-M. Copper-catalyzed synthesis od trifluoromethyl-substituted isoxazolines. Chem. Commun. 2013, 49, 5687–5689.
- Gao, P.; Yan, X.-B.; Tao, T.; Yang, F.; He, T.; Song, Z.-R.; Liu, X.-L.; Liang, Y.-M. Copper-Catalyzed Trifluoromethylation-Cyclization of Enynes: Highly Regioselective Construction of Trifluoromethylated Carbocycles and Heterocycles. Chem. Eur. J. 2013, 19, 14420–14424.
- Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Alkene Trifluoromethylation Coupled with C-C Bond Formation: Con-struction of Trifluoromethylated Carbocycles and Heterocycles. Angew. Chem. Int. Ed. 2013, 52, 4000–4003.
- Lin, J.-S.; Xiong, Y.-P.; Ma, C.-L.; Zhao, L.-J.; Tan, B.; Liu, X.-Y. Efficient Copper-Catalyzed Direct Intramolecular Aminotrifluoromethylation of Unactivated Alkenes with Diverse Nitrogen-Based Nucleophiles. Chem. Eur. J. 2014, 20, 1332–1340.
- Zhou, T.; Chen, Z.-C. Hypervalent iodine in synthesis. 77. An efficient method for the synthesis of N-arylindoles by the cop-per-catalyzed N-arylation of indole with diaryliodonium salts. Synth. Commun. 2002, 32, 903–907.
- Zhou, T.; Chen, Z.-C. Hypervalent Iodine in Synthesis 85: An Efficient Method for the Synthesis of N-Arylbenzimidazoles by the Copper- Catalyzed N-Arylation of Benzimidazole with Diaryliodonium Salts. Heteroat. Chem. 2002, 13, 617–619.
- Zhou, T.; Chen, Z.C. Hypervalent iodine in synthesis 87: The synthesis of 2,5-diaryl-2H-tetrazoles. J. Chem. Res. 2004, 2004, 404–405.
- Zhou, T.; Li, T.C.; Chen, Z.-C. Hypervalent Iodine in Synthesis. Part 86. Helv. Chim. Acta 2005, 88, 290–296.
- Saikia, R.A.; Barman, D.; Dutta, A.; Thakur, A.J. N1- and N3-Arylations of Hydantoins Employing Diaryliodonium Salts via Copper(I) Catalysis at Room Temperature. Eur. J. Org. Chem. 2021, 2021, 400–410.
- Geng, X.; Mao, S.; Chen, L.; Yu, J.; Han, J.; Hua, J.; Wang, L. Copper-catalyzed direct N-arylsulfonamides using diaryliodonium salts in water. Tetrahedron Lett. 2014, 55, 3856–3859.
- Vaddula, B.; Leazer, J.; Varma, R.S. Copper-Catalyzed Ultrasound-Expedited N-Arylation of Sulfoximines using Diaryliodonium Salts. Adv. Synth. Catal. 2012, 354, 986–990.
- Watanabe, K.; Moriyama, K. Copper-Catalyzed Indole-Selective C–N Coupling Reaction of Indolyl(2-alkoxy-phenyl)iodonium Imides: Effect of Substituent on Iodoarene as Dummy Ligand. J. Org. Chem. 2018, 83, 14827–14833.
- Watanabe, K.; Moriyama, K. Cu-Catalyzed Oxidative 3-Amination of Indoles via Formation of Indolyl(aryl)iodonium Imides Using o-Substituted (Diacetoxyiodo)arene as a High-Performance Hypervalent Iodine Compound. Molecules 2019, 24, 1147.
- Sokolovs, I.; Lubriks, D.; Suna, E. Copper-Catalyzed Intermolecular C–H Amination of (Hetero)arenes via Transient Unsymmetrical λ3-Iodanes. J. Am. Chem. Soc. 2014, 136, 6920–6928.
- Berzina, B.; Sokolovs, I.; Suna, E. Copper-Catalyzed para-Selective C–H Amination of Electron-Rich Arenes. ACS Catal. 2015, 5, 7008–7014.
- Wu, Y.; Izquierdo, S.; Vidossich, P.; Lledýs, A.; Shafir, A. NH-Heterocyclic Aryliodonium Salts and their Selective Conversion into N1-Aryl-5-iodoimidazoles. Angew. Chem. Int. Ed. 2016, 55, 7152–7156.
- Lv, T.; Wang, Z.; You, J.; Lan, J.; Gao, G. Copper-Catalyzed Direct Aryl Quaternization of N-Substituted Imidazoles to Form Imidazolium Salts. J. Org. Chem. 2013, 78, 5723–5730.
- Reus, C.; Stolar, M.; Vanderkley, J.; Nebauer, J.; Baumgartner, T. A Convenient N-Arylation Route for Electron-Deficient Pyridines: The Case of π-Extended Electrochromic Phosphaviologens. J. Am. Chem. Soc. 2015, 137, 11710–11717.
- Li, P.; Cheng, G.; Zhang, H.; Xu, X.; Gao, J.; Cui, X. Copper-Catalyzed One-Pot Synthesis of Unsymmetrical Arylurea Derivatives via Tandem Reaction of Diaryliodonium Salts with N-Arylcyanamide. J. Org. Chem. 2014, 79, 8156–8162.
- Peng, X.; Sun, Z.; Kuang, P.; Li, L.; Chen, J.; Chemn, J. Copper-Catalyzed Selective Arylation of Nitriles with Cyclic Diaryl Iodonium Salts: Direct Access to Structurally Diversified Diarylmethane Amides with Potential Neuroprotective and Anticancer Activities. Org. Lett. 2020, 22, 5789–5795.
- Aradia, K.; Novak, Z. Copper-Catalyzed Oxidative Ring Closure of ortho-Cyanoanilides with Hypervalent Iodonium Salts: Arylation–Ring Closure Approach to Iminobenzoxazines. Adv. Synth. Catal. 2015, 357, 371–376.
- Wang, Y.; Chen, C.; Peng, J.; Li, J. Copper(II)-Catalyzed Three-Component Cascade Annulation of Diaryliodoniums, Nitriles, and Alkynes: A Regioselective Synthesis of Multiply Substituted Quinolines. Angew. Chem. Int. Ed. 2013, 52, 5323–5327.
- Zhang, Y.; Lu, J.; Lan, T.; Cheng, S.; Liu, W.; Chen, C. Preparation, Characterization, and Reactivity of Aliphatic Amino Iodane(III) Reagents. Eur. J. Org. Chem. 2021, 2021, 436–442.
- Tianlei, L.; Yue, Z.; Wei, L.; Chanjuan, X.; Chao, C. Carbazolation Study of Active Arenes with Carbazole-Containing Hypervalent Iodine(III) Reagents. Chin. J. Org. Chem. 2019, 39, 2166–2174.
- Zhu, D.; Liu, Q.; Luo, B.; Chen, M.; Pi, R.; Huang, P.; Wen, S. Synthesis of Carbazoles via One-Pot Copper-Catalyzed Amine Insertion into Cyclic Diphenyleneiodoniums as a Strategy to Generate a Drug-Like Chemical Library. Adv. Synth. Catal. 2013, 355, 2172–2178.
- Xu, S.; Zhao, K.; Gu, Z. Copper-Catalyzed Asymmetric Ring-Opening of Cyclic Diaryliodonium with Benzylic and Aliphatic Amines. Adv. Synth. Catal. 2018, 360, 3877–3883.
- Zhang, X.; Zhao, K.; Li, N.; Yu, J.; Gong, L.-Z.; Gu, Z. Atroposelective Ring Opening of Cyclic Diaryliodonium Salts with Bulky Anilines Controlled by a Chiral Cobalt(III) Anion. Angew. Chem. Int. Ed. 2020, 59, 19899–19904.
- Li, Q.; Zhang, M.; Zhan, S.; Gu, Z. Copper-Catalyzed Enantioselective Ring-Opening of Cyclic Diaryliodoniums and O-Alkylhydroxylamines. Org. Lett. 2019, 21, 6374–6377.
- Chao, Z.; Ma, M.; Gu, Z. Cu-Catalyzed Site-Selective and Enantioselective Ring Opening of Cyclic Diaryliodoniums with 1,2,3-Triazoles. Org. Lett. 2020, 22, 6441–6446.
- Guo, W.; Gu, J.; Gu, Z. Catalytic Asymmteric Synthesis of Atropisomeric Nitrones: Versatile Intermediate for Axially Chiral Biaryls. Org. Lett. 2020, 22, 7622–7628.
- Hu, Y.; Zheng, S.; Fan, W.; Yuana, W. Copper-Catalysed Electrophilic Amination of Aryl(alkenyl) Boronic Acids with Nitrogen-Containing Hypervalent Iodine (III) Reagent. Adv. Synth. Catal. 2021, 363, 4701–4707.
- Tanaka, K.; Yoshida, M.; Yamamoto, A.; Hashimoto, Y.; Morita, N.; Tamura, O. Synthesis of N-Aryl Isoxazolidines by Photo-Promoted N-Selective Arylation of Oximes and Cyclization Using Hypervalent Iodine Reagents and Copper Catalyst. Adv. Synth. Catal. 2023, 365, 1419–1424.
- Sivaguru, P.; Ning, Y.; Bi, X. New Strategies for the Synthesis of Aliphatic Azides. Chem. Rev. 2021, 121, 4253–4307.
- Sala, R.; Loro, C.; Foschi, F.; Broggini, G. Transition Metal Catalyzed Azidation Reactions. Catalysts 2020, 10, 1173.
- Foschi, F.; Loro, C.; Sala, R.; Oble, J.; Lo Presti, L.; Beccalli, E.M.; Poli, G.; Broggini, G. Intramolecular Aminoazidation of Unactivated Terminal Alkenes by Palladium-Catalyzed Reactions with Hydrogen Peroxide as the Oxidant. Org. Lett. 2020, 22, 1402–1406.
- Lubriks, D.; Sokolovs, I.; Suna, E. Indirect C–H Azidation of Heterocycles via Copper-Catalyzed Regioselective Fragmentation of Unsymmetrical λ3-Iodanes. J. Am. Chem. Soc. 2012, 134, 15436–15442.
- Fan, Y.; Wan, W.; Ma, G.; Gao, W.; Jiang, H.; Zhuc, S.; Hao, J. Room-Temperature Cu(ii)-Catalyzed Aromatic C–H Azidation for the Synthesis of Ortho-Azido Anilines with Excellent Regioselectivity. Chem. Commun. 2014, 50, 5733–5736.
- Chen, Y.-X.; Huo, T.; Yin, Q.; Jiang, L.-F.; Cheng, X.; Ma, H.-X.; Jiang, Y.-X.; Sun, M.-Z.; Deng, Q.-H. Azidobenziodazolones as Azido Sources for the Enantioselective Copper-Catalyzed Azidation of N-Unprotected 3-Trifluoromethylated Oxindoles. Org. Lett. 2023, 25, 2739–2744.
- Yin, H.; Wang, T.; Jiao, N. Copper-Catalyzed Oxoazidation and Alkoxyazidation of Indoles. Org. Lett. 2014, 16, 2302–2305.
- Fumagalli, G.; Rabet, P.T.G.; Boyd, S.; Greaney, M.F. Three-Component Azidation of Styrene-Type Double Bonds: Light-Switchable Behavior of a Copper Photoredox Catalyst. Angew. Chem. Int. Ed. 2015, 54, 11481–11484.
- Rabet, P.T.R.; Fumagalli, G.; Boyd, S.; Greaney, M.F. Benzylic C–H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett. 2016, 18, 1646–1649.
- Shen, K.; Wang, Q. Copper-Catalyzed Alkene Aminoazidation as a Rapid Entry to 1,2-Diamines and Installation of an Azide Reporter onto Azahetereocycles. J. Am. Chem. Soc. 2017, 139, 13110–13116.
- Sokolovs, I.; Suna, E. Para-Selective Cu-Catalyzed C–H Aryloxylation of Electron-Rich Arenes and Heteroarenes. J. Org. Chem. 2016, 81, 371–379.
- Bhattarai, B.; Tay, J.-H.; Nagorny, P. Thiophosphoramides as Cooperative Catalysts for Copper-Catalyzed Arylation of Carboxylates with Diaryliodonium Salts. Chem. Commun. 2015, 51, 5398–5401.
- Xie, H.; Yang, S.; Zhang, C.; Ding, M.; Liu, M.; Guo, J.; Zhang, F. Copper-Catalyzed Selective Diphenylation of Carboxylic Acids with Cycli Diaryliodonium Salts. J. Org. Chem. 2017, 82, 5250–5262.
- Zhu, K.; Xu, K.; Fang, Q.; Wang, Y.; Tang, B.; Zhang, F. Enantioselective Synthesis of Axially Chiral Biaryls via Cu-Catalyzed Acyloxylation of Cyclic Diaryliodonium Salts. ACS Catal. 2019, 9, 4951–4957.
- Fañanás-Mastral, M.; Feringa, B.L. Copper-Catalyzed Synthesis of Mixed Alkyl Aryl Phosphonates. J. Am. Chem. Soc. 2014, 136, 9894–9897.
- Wang, G.; Xiong, B.; Zhou, C.; Liu, C.; Xu, W.; Yang, C.-A.; Tang, K.-W.; Wong, W.-Y. Copper-Catalyzed Diphenylation of P(O)-OH Bonds with Cyclic Diaryliodonium Salts. Chem. Asian J. 2019, 14, 4365–4374.
- Duan, L.; Zhao, K.; Wang, Z.; Zhang, F.-L.; Gu, Z. Enantioselective Ring-Opening/Oxidative Phosphorylation and P-Transfer Reaction of Cyclic Diaryliodoniums. ACS Catal. 2019, 9, 9852–9858.
- Vázquez-Galiñanes, N.; Andón-Rodríguez, M.; Gómez-Roibás, P.; Fañanás-Mastral, M. Copper-catalyzed O-alkenylation of phosphonates. Beilstein J. Org. Chem. 2020, 16, 611–615.
- Suero, M.G.; Bayle, E.D.; Collins, B.S.L.; Gaunt, M.J. Copper-Catalyzed Electrophilic Carbofunctionalization of Alkynes to Highly Functionalized Tetrasubstituted Alkenes. J. Am. Chem. Soc. 2013, 135, 5332–5335.
- Zhu, D.; Sun, Y.; Hui, P.; Li, H.; Yan, Y.; Kuang, H. Enantioselective Synthesis of Axially Chiral Oxazole Biaryls by Copper-Catalyzed Oxidation of Cyclic Diaryliodoniums. Eur. J. Org. Chem. 2022, 2022, e202101449.
- Zhu, D.; Liu, P.; Lu, W.; Wang, H.; Luo, B.; Hu, Y.; Huang, P.; Wen, S. Relayed Regioselective Alkynylation/Olefination of Unsymmetrical Cyclic Diaryliodonium Species Catalyzed by Cu and Pd: Affording Fluorescent Cytotoxic Benzoxazoles. Chem. Eur. J. 2015, 21, 18915–18920.
- Zhu, D.; Wu, Z.; Luo, B.; Du, Y.; Liu, P.; Chen, Y.; Hu, Y.; Huang, P.; Wen, S. Heterocyclic Iodoniums for the Assembly of Oxygen-Bridged Polycyclic Heteroarenes with Water as the Oxygen Source. Org. Lett. 2018, 20, 4815–4818.
- Zhu, D.; Li, M.; Wu, Z.; Du, Y.; Luo, B.; Huang, P.; Wen, S. Copper-Catalyzed One-Pot Synthesis of Dibenzofurans, Xanthenes, and Xanthones from Cyclic Diphenyl Iodoniums. Eur. J. Org. Chem. 2019, 2019, 4566–4571.
- Li, J.; Xu, Q.-N.; Wang, Z.-B.; Li, Y.; Liu, L. Synthesis of Dibenzofurans from Cyclic Diaryliodonium Triflates and Water via Oxygen–Iodine Exchange Approach. ACS Omega 2018, 3, 12923–12929.
- Luo, B.; Cui, Q.; Luo, H.; Hu, Y.; Huang, P.; Wen, S. N-Benzyldithiocarbamate Salts as Sulfur Sources to Access Tricyclic Thioheterocycles Mediated by Copper Species. Adv. Synth. Catal. 2016, 358, 2733–2738.
- Wang, M.; Wei, J.; Fan, Q.; Jiang, X. Cu(II)-catalyzed sulfide construction: Both aryl groups utilization of intermolecular and intramolecular diaryliodonium salt. Chem. Commun. 2017, 53, 2918–2921.
- Zhang, X.; Zhao, K.; Gu, Z. Transition Metal-Catalyzed Biaryl Atropisomer Synthesis via a Torsional Strain Promoted Ring-Opening Reaction. Acc. Chem. Res. 2022, 55, 1620–1633.
- Hou, M.; Deng, R.; Gu, Z. Cu-Catalyzed Enantioselective Atropisomer Synthesis via Thiolative Ring Opening of Five-Membered Cyclic Diaryliodoniums. Org. Lett. 2018, 20, 5779–5783.
- Li, B.; Chao, Z.; Li, C.; Gu, Z. Cu-Catalyzed Enantioselective Ring Opening of Cyclic Diaryliodoniums toward the Synthesis of Chiral Diarylmethanes. J. Am. Chem. Soc. 2018, 140, 9400–9403.
- Cullen, S.C.; Shekhar, S.; Nere, N.K. Cu-Catalyzed Couplings of Aryl Iodonium Salts with Sodium Trifluoromethanesulfinate. J. Org. Chem. 2013, 78, 12194–12201.
- Borrel, J.; Pisella, G.; Waser, J. Copper-Catalyzed Oxyalkynylation of C–S Bonds in Thiiranes and Thiethanes with Hypervalent Iodine Reagents. Org. Lett. 2020, 22, 422–427.
- Wang, M.; Chen, S.; Jiang, X. Construction of Functionalized Annulated Sulfone via SO2/I Exchange of Cyclic Diaryliodonium Salts. Org. Lett. 2017, 19, 4916–4919.
- Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. Trifluoromethanesulfonyl Hypervalent Iodonium Ylide for Copper-Catalyzed Trifluoromethylthiolation of Enamines, Indoles, and β-Keto Esters. J. Am. Chem. Soc. 2013, 135, 8782–8785.
- Duan, L.; Wang, Z.; Zhao, K.; Gu, Z. Enantioselective preparation of atropisomeric biaryl trifluoromethylsulfanes via ring-opening of cyclic diaryliodoniums. Chem. Commun. 2021, 57, 3881.
- Khan, S.; Volla, C.M.R. Cu-catalyzed Cascade Cyclization of Isothiocyanates, Alkynes, and Diaryliodonium Salts: Access to Diversely Functionalized Quinolines. Chem. Eur. J. 2017, 23, 12462–12466.
- Wang, W.; Zhou, J.; Wang, C.; Zhang, C.; Zhang, X.-Q.; Wang, Y. Design, development and applications of copper-catalyzed regioselective (4+2) annulations between diaryliodonium salts and alkynes. Commun. Chem. 2022, 5, 145.
- Liu, Z.; Zhu, D.; Luo, B.; Zhang, N.; Liu, Q.; Hu, Y.; Pi, R.; Huang, P.; Wen, S. Mild Cu(I)-Catalyzed Cascade Reaction of Cyclic Diaryliodoniums, Sodium Azide, and Alkynes: Efficient Synthesis of Trazolophenanthridines. Org. Lett. 2014, 16, 5600–5603.
- Urones, B.; Martínez, A.M.; Rodríguez, N.; Arrayás, R.G.; Carretero, J.C. Copper-catalyzed ortho-halogenation of protected anilines. Chem. Commun. 2013, 49, 11044–11046.
- Giofré, S.; Loro, C.; Molteni, L.; Castellano, C.; Contini, A.; Nava, D.; Broggini, G.; Beccalli, E.M. Copper(II)-Catalyzed Aminohalogenation of Alkynyl Carbamates. Eur. J. Org. Chem. 2021, 2021, 1750–1757.
- Hao, W.; Liu, Y. C-H bond halogenation catalyzed or mediated by copper: An overview. Beilstein J. Org. Chem. 2015, 11, 2132–2144.
- Yuan, W.; Szabò, K.J. Catalytic Intramolecular Aminofluorination, Oxyfluorination, and Carbofluorination with a Stable and Versatile Hypervalent Fluoroiodine Reagent. Angew. Chem. Int. Ed. 2015, 54, 8533–8537.
- Parvathaneni, S.P.; Perumgani, P.C. Regioselective Chlorination of Aryl C-H bonds with Hypervalent Iodine(III) Reagent 1-Chloro-1,2-benziodoxol-3-one. Asian J. Org. Chem. 2018, 7, 324–327.
- Wu, B.; Yoshikai, N. Conversion of 2-Iodobiaryls into 2,2’-Diiodobiaryls via Oxidation- Iodination Sequences: A Versatile Route to Ladder-Type Heterofluorenes. Angew. Chem. Int. Ed. 2015, 54, 8736–8739.
- Ke, J.; Zu, B.; Guo, Y.; Li, Y.; He, C. Hexafluoroisopropanol-Enabled Copper-Catalyzed Asymmetric Halogenation of Cyclic Diaryliodoniums for the Synthesis of Axially Chiral 2,2’-Dihalobiaryls. Org. Lett. 2021, 23, 329–333.
- Xu, J.; Zhang, P.; Gao, Y.; Chen, Y.; Tang, G.; Zhao, Y. Copper-Catalyzed P-Arylation via Direct Coupling of Diaryliodonium Salts with Phosphorus Nucleophiles at Room Temperature. J. Org. Chem. 2013, 78, 8176–8183.
- Beaud, R.; Phipps, R.J.; Gaunt, M.J. Enantioselective Cu-Catalyzed Arylation of Secondary Phosphine Oxides with Diaryliodonium Salts toward the Synthesis of P-Chiral Phosphines. J. Am. Chem. Soc. 2016, 138, 13183–13186.
- Cho, S.H.; Yoon, J.; Chang, S. Intramolecular Oxidative C-N Bond for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions. J. Am. Chem. Soc. 2011, 133, 5996–6005.
- Mirallai, S.I.; Koutentis, P.A. The Conversion of 4-Anilinquinazoline- and 3-Aryl-4-imino-3,4-dihydro-quinazoline-2-carbonitriles into Benzoimidazoquinazoline-6-carbonitriles via Oxidative and Nonoxidative C-N Couplings. J. Org. Chem. 2015, 80, 8329–8340.
- Liu, Z.-J.; Lu, X.; Wang, G.; Li, L.; Jiang, W.-T.; Wang, Y.-D.; Xiao, B.; Fu, Y. Directing Group in Decarboxylative Cross-Coupling: Copper-Catalyzed Site-Selective C-N Bond Formation from Nonactivated Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2016, 138, 9714–9719.
- Yang, Y.-N.; Jiang, J.-L.; Shi, J. Mechanistic Study of Copper-Catalyzed Decarboxylative C-N Cross-Coupling with Hypervalent Iodine Oxidant. Organometallics 2017, 36, 2081–2087.
- Yuan, J.; Rao, C.B.; Liang, Y.; Zhang, R.; Zhang, Q.; Hou, L.; Dong, D. Copper-Catalyzed Regioselective Oxidation Cycloamidation of α--Alkylamides: Synthetic Route to Substituted Pyrrolidine-2,4—Diones. Adv. Synth. Catal. 2019, 361, 160–169.
- Moreau, B.; Alberico, D.; Lindsay, V.N.G.; Charette, A.B. Catalytic asymmetric synthesis of nitrocyclopropane carboxylates. Tetrahedron 2012, 68, 3487–3496.
- Chidley, T.; Murphy, G.K. Cyclopropanation of alkenes with metallocarbenes generated from monocarbonyl iodonium ylides. Org. Biomol. Chem. 2018, 16, 8486–8490.
- Wan, J.-P.; Lin, Y.; Cao, X.; Liu, Y.; Wei, L. Copper-catalyzed, hypervalent iodine mediated C=C bond activation of enaminones for the synthesis of α-keto-amides. Chem. Commun. 2016, 52, 1270–1273.
- Loro, C.; Molteni, L.; Papis, M.; Beccalli, E.M.; Nava, D.; Lo Presti, L.; Brenna, S.; Colombo, G.; Foschi, F.; Broggini, G. Direct Synthesis of Fluorescent Oxazolo-phenoxazines by Copper-Catalyzed/Hypervalent Iodine(III)-Mediated Dimerization/Cyclization of 2-Benzylamino-phenols. J. Org. Chem. 2022, 87, 1032–1042.
- Liu, Z.; Huang, F.; Lou, J.; Wang, Q.; Yu, Z. Copper-promoted direct C-H alkoxylation of S,S-functionalized internal olefins with alcohols. Org. Biomol. Chem. 2017, 15, 5535–5540.
More
Information
Subjects:
Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
3 times
(View History)
Update Date:
29 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No