 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stefano Bettati | -- | 4224 | 2023-11-02 15:41:32 | | | |
| 2 | Lindsay Dong | Meta information modification | 4224 | 2023-11-06 01:58:47 | | |
Video Upload Options
The intracellular concentrations of oxygen and reactive oxygen species (ROS) in living cells represent critical information for investigating physiological and pathological conditions. Real-time measurement often relies on genetically encoded proteins that are responsive to fluctuations in either oxygen or ROS concentrations. The direct binding or chemical reactions that occur in their presence either directly alter the fluorescence properties of the binding protein or alter the fluorescence properties of fusion partners, mostly consisting of variants of the green fluorescent protein. Oxygen sensing takes advantage of several mechanisms, including (i) the oxygen-dependent hydroxylation of a domain of the hypoxia-inducible factor-1, which, in turn, promotes its cellular degradation along with fluorescent fusion partners; (ii) the naturally oxygen-dependent maturation of the fluorophore of green fluorescent protein variants; and (iii) direct oxygen binding by proteins, including heme proteins, expressed in fusion with fluorescent partners, resulting in changes in fluorescence due to conformational alterations or fluorescence resonance energy transfer.
1. Relevance of O2 and ROS Sensing
2. Biosensors for O2
2.1. O2 Sensing Mediated by Prolyl Hydroxylases
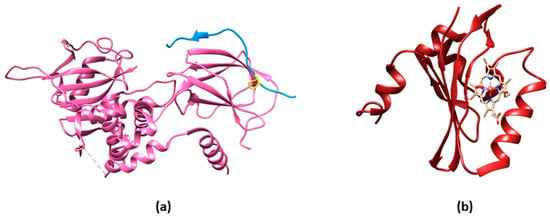
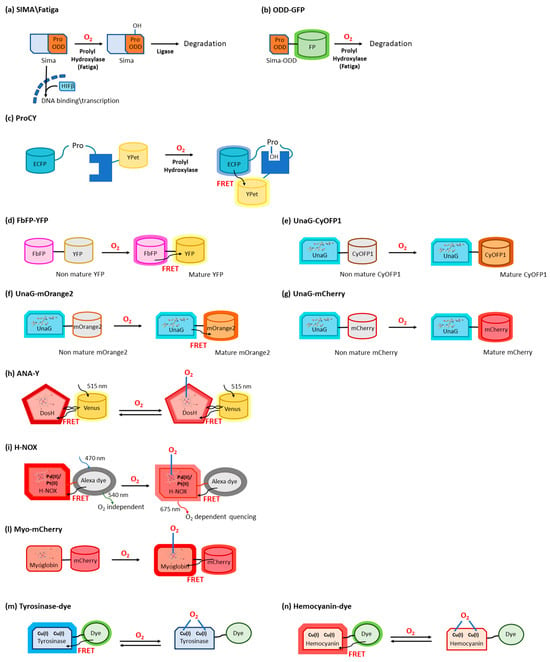
2.2. O2 Sensors Based on O2-Dependent Maturation of GFP Variants
2.3. O2 Sensors Based on O2-Binding Heme Proteins
2.4. O2 Sensors Based on O2-Binding Copper Proteins
3. Biosensors for ROS
3.1. ROS Sensors Based on roFPs
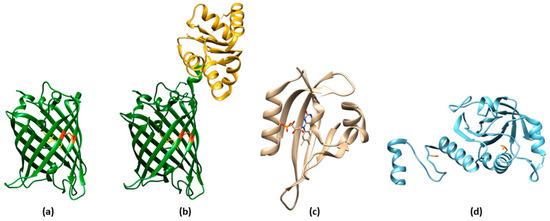
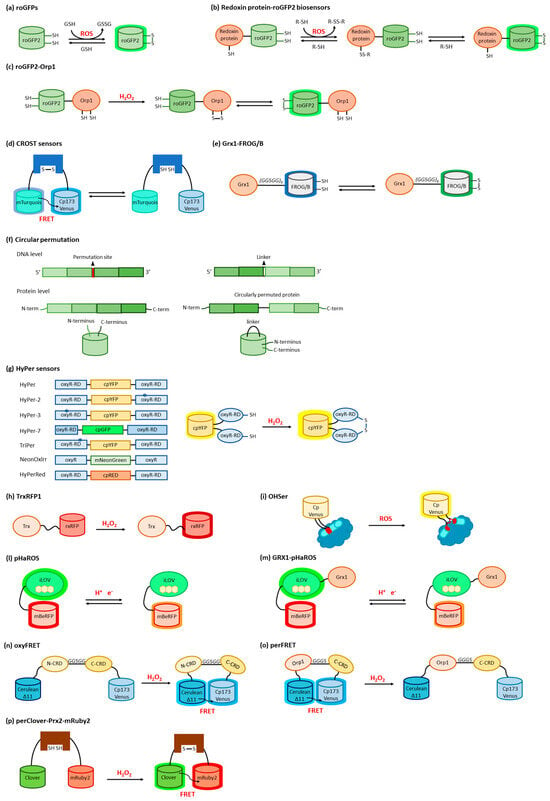
3.2. ROS Sensors Based on roGFP Fusion Proteins
3.3. ROS Sensors Based on Circularly Permuted FPs
3.4. ROS Sensors Based on LOV Domains
3.5. ROS Sensors Based on YAP1
3.6. ROS Sensors Based on Peroxiredoxin
References
- Raymond, J.; Segrè, D. The Effect of Oxygen on Biochemical Networks and the Evolution of Complex Life. Science 2006, 311, 1764–1767.
- Della Rocca, Y.; Fonticoli, L.; Rajan, T.S.; Trubiani, O.; Caputi, S.; Diomede, F.; Pizzicannella, J.; Marconi, G.D. Hypoxia: Molecular pathophysiological mechanisms in human diseases. J. Physiol. Biochem. 2022, 78, 739–752.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Görlach, A.; Camenisch, G.; Kvietikova, I.; Vogt, L.; Wenger, R.H.; Gassmann, M. Efficient translation of mouse hypoxia-inducible factor-1α under normoxic and hypoxic conditions. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1493, 125–134.
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF–1α in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314.
- Bruick, R.K.; McKnight, S.L. Oxygen Sensing Gets a Second Wind. Science 2002, 295, 807–808.
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J. Biol. Chem. 1997, 272, 11205–11214.
- Masson, N.; Ratcliffe, P.J. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J. Cell Sci. 2003, 116, 3041–3049.
- Hon, W.-C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.W.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 2002, 417, 975–978.
- Park, H.; Suquet, C.; Satterlee, J.D.; Kang, C. Insights into Signal Transduction Involving PAS Domain Oxygen-Sensing Heme Proteins from the X-ray Crystal Structure of Escherichia Coli Dos Heme Domain (Ec DosH). Biochemistry 2004, 43, 2738–2746.
- Arquier, N.; Vigne, P.; Duplan, E.; Hsu, T.; Therond, P.P.; Frelin, C.; D’Angelo, G. Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem. J. 2005, 393, 471–480.
- Lavista-Llanos, S.; Centanin, L.; Irisarri, M.; Russo, D.M.; Gleadle, J.M.; Bocca, S.N.; Muzzopappa, M.; Ratcliffe, P.J.; Wappner, P. Control of the Hypoxic Response in Drosophila melanogaster by the Basic Helix-Loop-Helix PAS Protein Similar. Mol. Cell. Biol. 2002, 22, 6842–6853.
- Bacon, N.C.M.; Wappner, P.; O’Rourke, J.F.; Bartlett, S.M.; Shilo, B.; Pugh, C.W.; Ratcliffe, P.J. Regulation of the Drosophila bHLH-PAS Protein Sima by Hypoxia: Functional Evidence for Homology with Mammalian HIF-1α. Biochem. Biophys. Res. Comm. 1998, 249, 811–816.
- Youssef, S.; Ren, W.; Ai, H.-w. A Genetically Encoded FRET Sensor for Hypoxia and Prolyl Hydroxylases. ACS Chem. Biol. 2016, 11, 2492–2498.
- Remington, S.J. Fluorescent proteins: Maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006, 16, 714–721.
- Chapman, S.; Faulkner, C.; Kaiserli, E.; Garcia-Mata, C.; Savenkov, E.I.; Roberts, A.G.; Oparka, K.J.; Christie, J.M. The photoreversible fluorescent protein iLOV outperforms GFP as a reporter of plant virus infection. Proc. Natl. Acad. Sci. USA 2008, 105, 20038–20043.
- Drepper, T.; Eggert, T.; Circolone, F.; Heck, A.; Krauß, U.; Guterl, J.-K.; Wendorff, M.; Losi, A.; Gärtner, W.; Jaeger, K.-E. Reporter proteins for in vivo fluorescence without oxygen. Nat. Biotech. 2007, 25, 443–445.
- Mukherjee, A.; Walker, J.; Weyant, K.B.; Schroeder, C.M. Characterization of Flavin-Based Fluorescent Proteins: An Emerging Class of Fluorescent Reporters. PLoS ONE 2013, 8, e64753.
- Delvigne, F.; Boxus, M.; Ingels, S.; Thonart, P. Bioreactor mixing efficiency modulates the activity of a prpoS::GFP reporter gene in E. coli. Microb. Cell Factories 2009, 8, 15.
- Garcia, J.R.; Cha, H.J.; Rao, G.; Marten, M.R.; Bentley, W.E. Microbial nar-GFP cell sensors reveal oxygen limitations in highly agitated and aerated laboratory-scale fermentors. Microb. Cell Factories 2009, 8, 6.
- Potzkei, J.; Kunze, M.; Drepper, T.; Gensch, T.; Jaeger, K.-E.; Büchs, J. Real-time determination of intracellular oxygen in bacteria using a genetically encoded FRET-based biosensor. BMC Biol. 2012, 10, 28.
- Bauer, N.; Maisuls, I.; Pereira da Graça, A.; Reinhardt, D.; Erapaneedi, R.; Kirschnick, N.; Schäfers, M.; Grashoff, C.; Landfester, K.; Vestweber, D.; et al. Genetically encoded dual fluorophore reporters for graded oxygen-sensing in light microscopy. Biosens. Bioelectron. 2023, 221, 114917.
- Panicucci, G.; Iacopino, S.; De Meo, E.; Perata, P.; Weits, D.A. An Improved HRPE-Based Transcriptional Output Reporter to Detect Hypoxia and Anoxia in Plant Tissue. Biosensors 2020, 10, 197.
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell 2013, 153, 1602–1611.
- Bettati, S.; Pasqualetto, E.; Lolli, G.; Campanini, B.; Battistutta, R. Structure and single crystal spectroscopy of Green Fluorescent Proteins. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 824–833.
- Sawai, H.; Yoshioka, S.; Uchida, T.; Hyodo, M.; Hayakawa, Y.; Ishimori, K.; Aono, S. Molecular oxygen regulates the enzymatic activity of a heme-containing diguanylate cyclase (HemDGC) for the synthesis of cyclic di-GMP. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 166–172.
- Shimizu, T. The Heme-Based Oxygen-Sensor Phosphodiesterase Ec DOS (DosP): Structure-Function Relationships. Biosensors 2013, 3, 211–237.
- Stranava, M.; Martínek, V.; Man, P.; Fojtikova, V.; Kavan, D.; Vaněk, O.; Shimizu, T.; Martinkova, M. Structural characterization of the heme-based oxygen sensor, AfGcHK, its interactions with the cognate response regulator, and their combined mechanism of action in a bacterial two-component signaling system. Proteins Struct. Funct. Bioinform. 2016, 84, 1375–1389.
- Tuckerman, J.R.; Gonzalez, G.; Sousa, E.H.S.; Wan, X.; Saito, J.A.; Alam, M.; Gilles-Gonzalez, M.-A. An Oxygen-Sensing Diguanylate Cyclase and Phosphodiesterase Couple for c-di-GMP Control. Biochemistry 2009, 48, 9764–9774.
- Park, H.; Suquet, C.; Savenkova, M.I.; Satterlee, J.D.; Kang, C. Cloning, purification, crystallization and preliminary X-ray analysis of DOS heme domain, a new heme oxygen sensor in Escherichia coli. Acta Crystallogr. D 2002, 58, 1504–1506.
- Ishitsuka, Y.; Araki, Y.; Tanaka, A.; Igarashi, J.; Ito, O.; Shimizu, T. Arg97 at the Heme-Distal Side of the Isolated Heme-Bound PAS Domain of a Heme-Based Oxygen Sensor from Escherichia coli (Ec DOS) Plays Critical Roles in Autoxidation and Binding to Gases, Particularly O2. Biochemistry 2008, 47, 8874–8884.
- Nomata, J.; Hisabori, T. Development of heme protein based oxygen sensing indicators. Sci. Rep. 2018, 8, 11849.
- Lemon, C.M.; Hanley, D.; Batka, A.E.; Marletta, M.A. Ratiometric Oxygen Sensing with H-NOX Protein Conjugates. Inorg. Chem. 2022, 61, 10521–10532.
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606.
- Zauner, G.; Lonardi, E.; Bubacco, L.; Aartsma, T.J.; Canters, G.W.; Tepper, A.W.J.W. Tryptophan-to-Dye Fluorescence Energy Transfer Applied to Oxygen Sensing by Using Type-3 Copper Proteins. Chem. Eur. J. 2007, 13, 7085–7090.
- Solomon, E.I.; Chen, P.; Metz, M.; Lee, S.-K.; Palmer, A.E. Oxygen Binding, Activation, and Reduction to Water by Copper Proteins. Angew. Chem. Int. Ed. 2001, 40, 4570–4590.
- Erker, W.; Sdorra, S.; Basché, T. Detection of Single Oxygen Molecules with Fluorescence-Labeled Hemocyanins. J. Am. Chem. Soc. 2005, 127, 14532–14533.
- Østergaard, H.; Henriksen, A.; Hansen, F.G.; Winther, J.R. Shedding light on disulfide bond formation: Engineering a redox switch in green fluorescent protein. Embo J. 2001, 20, 5853–5862.
- Håkansson, K.O.; Winther, J.R. Structure of glutaredoxin Grx1p C30S mutant from yeast. Acta Crystallogr. D Biol. Crystallogr. 2007, 63 Pt 3, 288–294.
- Christie, J.M.; Hitomi, K.; Arvai, A.S.; Hartfield, K.A.; Mettlen, M.; Pratt, A.J.; Tainer, J.A.; Getzoff, E.D. Structural Tuning of the Fluorescent Protein iLOV for Improved Photostability. J. Biol. Chem. 2012, 287, 22295–22304.
- Schröder, E.; Littlechil, J.A.; Lebedev, A.A.; Errington, N.; Vagin, A.A.; Isupov, M.N. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure 2000, 8, 605–615.
- Østergaard, H.; Tachibana, C.; Winther, J.R. Monitoring disulfide bond formation in the eukaryotic cytosol. J. Cell Biol. 2004, 166, 337–345.
- Björnberg, O.; Østergaard, H.; Winther, J.R. Mechanistic insight provided by glutaredoxin within a fusion to redox-sensitive yellow fluorescent protein. Biochemistry 2006, 45, 2362–2371.
- Morgan, B.; Sobotta, M.C.; Dick, T.P. Measuring E(GSH) and H2O2 with roGFP2-based redox probes. Free Radic. Biol. Med. 2011, 51, 1943–1951.
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559.
- Trautsch, I.; Heta, E.; Soong, P.L.; Levent, E.; Nikolaev, V.O.; Bogeski, I.; Katschinski, D.M.; Mayr, M.; Zimmermann, W.H. Optogenetic Monitoring of the Glutathione Redox State in Engineered Human Myocardium. Front. Physiol. 2019, 10, 272.
- Mohring, F.; Jortzik, E.; Becker, K. Comparison of methods probing the intracellular redox milieu in Plasmodium falciparum. Mol. Biochem. Parasitol. 2016, 206, 75–83.
- Hatori, Y.; Kubo, T.; Sato, Y.; Inouye, S.; Akagi, R.; Seyama, T. Visualization of the Redox Status of Cytosolic Glutathione Using the Organelle- and Cytoskeleton-Targeted Redox Sensors. Antioxidants 2020, 9, 129.
- Pal, R.; Basu Thakur, P.; Li, S.; Minard, C.; Rodney, G.G. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS ONE 2013, 8, e63989.
- Henríquez-Olguín, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to high-intensity interval training in skeletal muscle require NADPH oxidase 2. Redox Biol. 2019, 24, 101188.
- Bhaskar, A.; Chawla, M.; Mehta, M.; Parikh, P.; Chandra, P.; Bhave, D.; Kumar, D.; Carroll, K.S.; Singh, A. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 2014, 10, e1003902.
- Van Loi, V.; Antelmann, H. Method for measurement of bacillithiol redox potential changes using the Brx-roGFP2 redox biosensor in Staphylococcus aureus. MethodsX 2020, 7, 100900.
- Van Loi, V.; Harms, M.; Müller, M.; Huyen, N.T.T.; Hamilton, C.J.; Hochgräfe, F.; Pané-Farré, J.; Antelmann, H. Real-Time Imaging of the Bacillithiol Redox Potential in the Human Pathogen Staphylococcus aureus Using a Genetically Encoded Bacilliredoxin-Fused Redox Biosensor. Antioxid. Redox Signal 2017, 26, 835–848.
- Ebersoll, S.; Bogacz, M.; Günter, L.M.; Dick, T.P.; Krauth-Siegel, R.L. A tryparedoxin-coupled biosensor reveals a mitochondrial trypanothione metabolism in trypanosomes. Elife 2020, 9, e53227.
- Sugiura, K.; Nagai, T.; Nakano, M.; Ichinose, H.; Nakabayashi, T.; Ohta, N.; Hisabori, T. Redox sensor proteins for highly sensitive direct imaging of intracellular redox state. Biochem. Biophys. Res. Commun. 2015, 457, 242–248.
- Sugiura, K.; Yokochi, Y.; Fu, N.; Fukaya, Y.; Yoshida, K.; Mihara, S.; Hisabori, T. The thioredoxin (Trx) redox state sensor protein can visualize Trx activities in the light/dark response in chloroplasts. J. Biol. Chem. 2019, 294, 12091–12098.
- Sugiura, K.; Mihara, S.; Fu, N.; Hisabori, T. Real-time monitoring of the in vivo redox state transition using the ratiometric redox state sensor protein FROG/B. Proc. Natl. Acad. Sci. USA 2020, 117, 16019–16026.
- Heinemann, U.; Hahn, M. Circular permutation of polypeptide chains: Implications for protein folding and stability. Prog. Biophys. Mol. Biol. 1995, 64, 121–143.
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246.
- Topell, S.; Hennecke, J.; Glockshuber, R. Circularly permuted variants of the green fluorescent protein. FEBS Lett. 1999, 457, 283–289.
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286.
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.J.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A Genetically Encoded H2O2 Probe with Improved Performance for Ratiometric and Fluorescence Lifetime Imaging. ACS Chem. Biol. 2013, 8, 535–542.
- Markvicheva, K.N.; Bilan, D.S.; Mishina, N.M.; Gorokhovatsky, A.Y.; Vinokurov, L.M.; Lukyanov, S.; Belousov, V.V. A genetically encoded sensor for H2O2 with expanded dynamic range. Bioorg Med. Chem. 2011, 19, 1079–1084.
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Martínez Gache, S.A.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6.
- Lee, C.; Lee, S.M.; Mukhopadhyay, P.; Kim, S.J.; Lee, S.C.; Ahn, W.-S.; Yu, M.-H.; Storz, G.; Ryu, S.E. Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat. Struct. Mol. Biol. 2004, 11, 1179–1185.
- Fan, Y.; Makar, M.; Wang, M.X.; Ai, H.-W. Monitoring thioredoxin redox with a genetically encoded red fluorescent biosensor. Nat. Chem. Biol. 2017, 13, 1045–1052.
- Fan, Y.; Ai, H.W. Development of redox-sensitive red fluorescent proteins for imaging redox dynamics in cellular compartments. Anal. Bioanal. Chem. 2016, 408, 2901–2911.
- Simen Zhao, B.; Liang, Y.; Song, Y.; Zheng, C.; Hao, Z.; Chen, P.R. A Highly Selective Fluorescent Probe for Visualization of Organic Hydroperoxides in Living Cells. J. Am. Chem. Soc. 2010, 132, 17065–17067.
- Herrou, J.; Crosson, S. Function, structure and mechanism of bacterial photosensory LOV proteins. Nat. Rev. Microbiol. 2011, 9, 713–723.
- Gu, Y.Z.; Hogenesch, J.B.; Bradfield, C.A. The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 519–561.
- Sakai, T.; Kagawa, T.; Kasahara, M.; Swartz, T.E.; Christie, J.M.; Briggs, W.R.; Wada, M.; Okada, K. Arabidopsis nph1 and npl1: Blue light receptors that mediate both phototropism and chloroplast relocation. Proc. Natl. Acad. Sci. USA 2001, 98, 6969–6974.
- Christie, J.M.; Salomon, M.; Nozue, K.; Wada, M.; Briggs, W.R. LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): Binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. USA 1999, 96, 8779–8783.
- Crosson, S.; Rajagopal, S.; Moffat, K. The LOV domain family: Photoresponsive signaling modules coupled to diverse output domains. Biochemistry 2003, 42, 2–10.
- Ravikumar, Y.; Nadarajan, S.P.; Lee, C.S.; Rhee, J.K.; Yun, H.D. A New-Generation Fluorescent-Based Metal Sensor—iLOV Protein. J. Microbiol. Biotechnol. 2015, 25, 503–510.
- Zhao, H.; Zhang, Y.; Pan, M.; Song, Y.; Bai, L.; Miao, Y.; Huang, Y.; Zhu, X.; Song, C.-P. Dynamic imaging of cellular pH and redox homeostasis with a genetically encoded dual-functional biosensor, pHaROS, in yeast. J. Biol. Chem. 2019, 294, 15768–15780.
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; et al. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746.
- Enyedi, B.; Zana, M.; Donkó, Á.; Geiszt, M. Spatial and temporal analysis of NADPH oxidase-generated hydrogen peroxide signals by novel fluorescent reporter proteins. Antioxid. Redox Signal 2013, 19, 523–534.
- Langford, T.F.; Huang, B.K.; Lim, J.B.; Moon, S.J.; Sikes, H.D. Monitoring the action of redox-directed cancer therapeutics using a human peroxiredoxin-2-based probe. Nat. Commun. 2018, 9, 3145.

