Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yousef Hawsawi | -- | 2051 | 2023-10-21 00:53:46 | | | |
| 2 | Nahed Mahrous | Meta information modification | 2051 | 2023-10-21 23:06:52 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2051 | 2023-10-23 05:06:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mahrous, N.N.; Jamous, Y.F.; Almatrafi, A.M.; Fallatah, D.I.; Theyab, A.; Alanati, B.H.; Alsagaby, S.A.; Alenazi, M.K.; Khan, M.I.; Hawsawi, Y.M. Characterization, Therapy and Management of Alport Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/50628 (accessed on 24 July 2026).
Mahrous NN, Jamous YF, Almatrafi AM, Fallatah DI, Theyab A, Alanati BH, et al. Characterization, Therapy and Management of Alport Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/50628. Accessed July 24, 2026.
Mahrous, Nahed N., Yahya F. Jamous, Ahmad M. Almatrafi, Deema I. Fallatah, Abdulrahman Theyab, Bayan H. Alanati, Suliman A. Alsagaby, Munifa K. Alenazi, Mohammed I. Khan, Yousef M. Hawsawi. "Characterization, Therapy and Management of Alport Syndrome" Encyclopedia, https://encyclopedia.pub/entry/50628 (accessed July 24, 2026).
Mahrous, N.N., Jamous, Y.F., Almatrafi, A.M., Fallatah, D.I., Theyab, A., Alanati, B.H., Alsagaby, S.A., Alenazi, M.K., Khan, M.I., & Hawsawi, Y.M. (2023, October 20). Characterization, Therapy and Management of Alport Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/50628
Mahrous, Nahed N., et al. "Characterization, Therapy and Management of Alport Syndrome." Encyclopedia. Web. 20 October, 2023.
Copy Citation
Alport syndrome (AS) is a rare genetic disorder categorized by the progressive loss of kidney function, sensorineural hearing loss and eye abnormalities. It occurs due to mutations in three genes that encode for the alpha chains of type IV collagen. Globally, the disease is classified based on the pattern of inheritance into X-linked AS (XLAS), which is caused by pathogenic variants in COL4A5, representing 80% of AS. Autosomal recessive AS (ARAS), caused by mutations in either COL4A3 or COL4A4, represents 15% of AS. Autosomal dominant AS (ADAS) is rare and has been recorded in 5% of all cases due to mutations in COL4A3 or COL4A4.
Alport syndrome
type IV collagen
glomerular basement membrane
kidney disease
gene technology
1. Introduction
In 1875, Dickinson was the first scientist who reported the presence of familial inherited renal failure. In 1972, Arthur Cecil Alport was the first British physician who described symptoms of kidney and hearing health problems in families. He pointed out that females were less severely affected by deafness than males, although the disorder was more likely transmitted by females. Further cases were diagnosed, and AS was approved in 1961 as an eponym [1][2]. It has been observed that AS develops frequently in patients with hematuria (i.e., bloody urine), nerve deafness, edema, and hypertension [3].
Alport syndrome is significantly caused by mutations in one of three genes that code for type IV collagen alpha chains: COL4A3 (α3), COL4A4 (α4) and/or COL4A5 (α5) [4]. Those genes are involved in type IV collagen biosynthesis [5]. Type IV collagen mainly constitutes of the glomerular basement membrane (GBM), which represents ~50% of its total protein mass and is responsible for its stability. However, GBM is a thin extracellular matrix protein that functions as a per selective barrier to the passage of blood cells and proteins from the blood to the urinary tract [6] (Figure 1(3)). Changes in α-chains can give rise to dysfunctional GBM, leading to sensorineural deafness, abnormalities in several parts of the eyes, hematuria, proteinuria (i.e., excessive protein in urine) and eventual chronic kidney disease (CKD) [7][8].
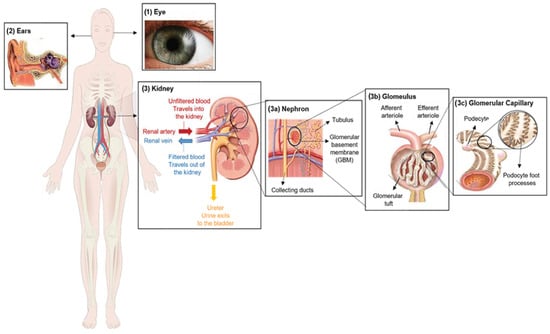
Figure 1. Clinical characteristics of Alport syndrome in individuals. Symptoms of Alport syndrome can be categorized by (1) eye abnormalities; (2) sensorineural hearing loss; and (3) related kidney symptoms.
Estimations of gene mutation frequency range from 1 in 5000 to 1 in 10,000, demonstrating its high burden in clinical settings [9]. As a rare disease, the estimated frequency of AS is approximately 1 in 50,000 live births worldwide [3]. Despite advances in therapeutic strategies that are performed to address many of the associated symptoms and slow the progression of kidney disease, there is no radical therapy for AS as of this moment.
Alport syndrome (AS) is a rare genetic disorder categorized by a progressive loss of kidney function, sensorineural hearing loss and eye abnormalities (Figure 1).
2. Clinical Characterization of Alport Syndrome
Clinically, AS characteristics are diverse, with a wide range progressing to end-stage renal disease (ESRD). The main and initial observed criterion among all the AS modes of inheritance is persistent hematuria, as approximately detected in all male and 97% of female patients [10]. Proteinuria is detected in male patients in early childhood, with levels gradually increasing with age, and occasionally presents as a nephrotic condition. It is also estimated that 90% of Proteinuric patients acquire ESRD by the age of 40 years, with the median age of ESRD development being 25 years. By the age of 40, 12% of females have acquired ESRD [10][11]. Also, Nozu et al. in 2019 reported that female XLAS patients developed proteinuria at a median age of 7 years and ESRD at a median age of 65 years [7]. Ultimately, 90% of male and 20% of female patients develop end-stage renal disease (ESRD) by the age of 40 years [11][12].
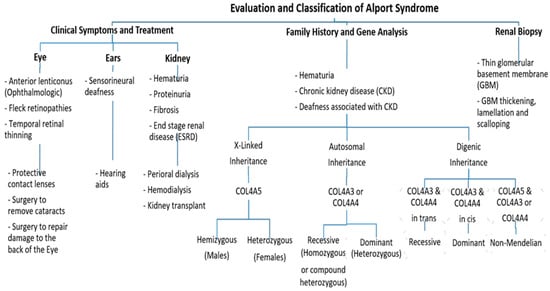
Further, ophthalmologic consequences include specific ocular abnormalities such as anterior lenticonus, posterior subcapsular cataract, posterior polymorphous dystrophy, and retinal flecks [13][14]. In addition, AS-affected patients frequently exhibit external disorders such as sensorineural deafness, of which frequently develops in late infancy; by the age of 40, 90% of male patients and around 12% of female patients have hearing loss [11][12] (Figure 2).
Figure 2. An approach regarding the evaluation and classification of Alport syndrome in individuals. In X-Linked Alport syndrome (X-Linked AS), the disease is usually passed from the mother to her child. In autosomal recessive Alport syndrome (ARAS), both sides of the family must have the mutation for AS to be passed on to their children, while in autosomal dominant Alport syndrome (ADAS), only one side of the family must have the mutation to pass it.
3. Genetics of Alport Syndrome
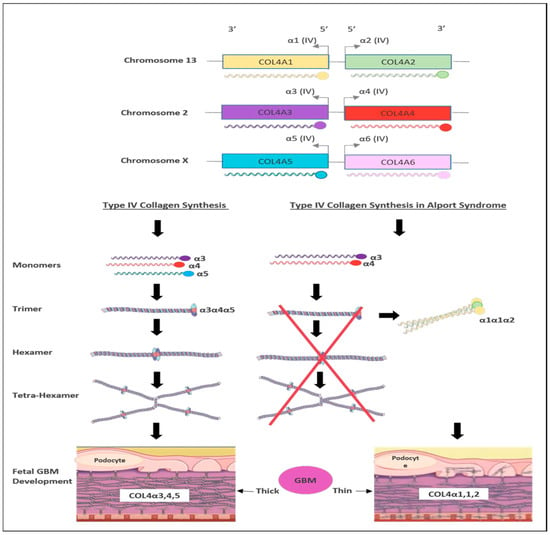
There are six members of type IV collagen (COL4A1, COL4A2, COL4A3, COL4A4, COL4A5 and COL4A6), which encode six unique alpha chains of type IV collagen (α1–α6) (Figure 3).
Figure 3. Schematic representation of type IV collagen biosynthesis. Type IV collagen genes are located in three different chromosomes pairwise, which encode six unique α-chains of type IV collagen (α1–α6). Monomers of collagen α3α4α5 can be combined among other forming triple helices (trimers). Two trimers then combine to form hexamers that can be associated with additional three hexamers that form tera-hexamer, which forms a thick GBM layer. However, in the absence of one monomer of the three α-chains, the kidney produces a monomer composed of α1α1α2 that eventually creates a thin GBM layer.
Pathogenic variants in these genes can cause several progressive and non-progressive glomerular disorders [15]. Mutations in COL4A5 are the main cause of XLAS (Figure 2) and are responsible for more than 80% of AS cases [4]. It is located on chromosome Xq22 and has approximately 51 exons that encode 1685 amino acids [16]. The protein structure of the α5 chain includes three parts: A signal peptide with 26 amino acid residues, a collagenous domain with 1430 residues that consist of short non-collagenous interruptions and a 229 amino acid carboxy-terminal non-collagenous (NC) domain [11].
On the other hand, mutations in COL4A3 or COL4A4 genes are the main cause of autosomal recessive AS (ARAS) or autosomal dominant AS (ADAS) (Figure 2). Those genes are arranged head-to-head on chromosome 2 (2q36.3) and both share one promotor that encodes other type IV collagen genes [17]. The COL4A3 gene consists of 52 exons that span over 88 kb and encode 1670 amino acids, whereas COL4A4 spans 161 kb and is composed of 48 exons [4]. Both COL4A3 and COL4A4 proteins are composed of the N-terminal amino acid domain, the central triple-helical domain including G-X-Y repeats, and the C-terminal globular non-collagenous domain with a highly homologous protein structure with COL4A5 [18].
The ARAS mutations in either COL4A3 or COL4A4 are responsible for 15% of AS patients with no family history of AS [19]. Individuals with ARAS exhibit clinical features that are similar to those seen in males with XLAS. Carriers with the monoallelic variant are frequently asymptomatic or show only mild proteinuria and microscopic hematuria [20]. However, individuals with truncating variants are more likely to develop kidney failure before the age of 30 [19][21].
The genotype–phenotype correlation has been studied in a European cohort [11]. Earlier genotype–phenotype correlation studies have revealed that males with large deletions, frameshift mutations, and truncating variants in the COL4A5 gene present with more severe clinical manifestations and are at a high risk of kidney failure at a younger age [11], whereas males with missense variants tend to exhibit milder disease features. In contrast, no clear genotype–phenotype correlation has been reported among females with heterozygous variants in COL4A5 and their age at kidney failure [12]. However, it has been observed that females with missense variants in the COL4A5 gene have better kidney function and are less likely to develop proteinuria in comparison to females carrying other types of variants [22]. A study by Bekheirnia et al. from 2010 examined the relationships between genotype and phenotype in a sizable US cohort of male patients with XLAS. As an outcome, they discovered that missense mutations (51% of the families) were most frequently found, followed by truncating (14%) and splice site (13.7%) alterations [13].
The most common pathogenic variants are the missense variants that affect Gly resides in the collagenous Gly Xaa Yaa repeats, which cause AS in Gly substitutions [4]. The clinical features and severity associated with Gly missense variants highly vary between patients [18]. In contrast, the mutation substitutions affecting non-collagenous boundary Gly residues resulted in kidney failure at a delayed age [4].
4. Current Therapies for Alport Syndrome Management
4.1. Chemical Drugs
According to the European Alport Registry, the renin angiotensin aldosterone system (RAAS) blocker is linked to a slower course of renal disease in heterozygous Alport carriers. Angiotensin-converting enzyme inhibitors (ACEIs) such as Ramipril can be used to treat heart failure and diabetic kidney diseases. An in vivo study showed that Ramipril with ARAS resulted in a significant reduction in various kidney diseases including proteinuria diseases [23].
Mineralocorticoid receptor antagonists (MRAs), such as finerenone, are widely used diuretics to reduce the severity of glomerulosclerosis, which is the hardening of the glomeruli in the kidney and renal interstitial fibrosis [24].
Sodium-Glucose Cotransporter-2 inhibitors (SGLT2i), such as Dapagliflozin, are FDA-approved drugs prescribed for patients who have type II diabetes. This treatment was considered to be immensely convenient in reducing the progression of chronic kidney failure and heart diseases [25]. A study of children with AS reported a 22% reduction in proteinuria after about three months of Dapagliflozin [26].
Metformin is a biguanide anti-hyperglycemic agent that can be used as first-line therapy to manage type II diabetes. It can reduce kidney fibrosis, inflammation and glomerular damage. The FDA has given Metformin a box warning and advises against using it in advanced renal disease (estimated glomerular filtration rate “eGFR” 30 mL/min/1.73 m2) due to its potential link to lactic acidosis [23].
Lipid-lowering agents such as statins are another treatment option. According to the Kidney Disease: Improving Global Outcomes (KDIGO) 2013 recommendations, people over 50 with an eGFR of less than 60 mL/min/1.73 m2 should take a statin. Patients with advanced renal disease are not covered by this (1A) [27]. It has been hypothesized that statin use may result in a slower course of renal disease [28].
4.2. Molecular Therapies
Short non-coding RNAs (<200 nucleotides) called microRNAs (miRNAs) can control the expression of genes by preventing or speeding up the degradation of the messenger RNAs they target [29]. Recently, several miRNAs have been introduced in clinical settings to be used for disease treatment, including miRNA-21. As well as other kidney diseases, the dysregulation of miRNA-21 has been discovered in AS. It was found that the use of anti-miRNA-21 oligonucleotides greatly slows the progression of kidney disease and increases survival in Alport mouse models after it was demonstrated that renal miRNA-21 is elevated in Col43/mice [30].
Several therapies improve outcomes in the animal models of AS including inhibitors of TGF-β1, vasopeptidase A and matrix metalloproteinases [31], BMP-7 [32], chemokine receptor 1 blockade [33], and stem cells [34]. However, none of these therapies have been prospectively researched in human AS populations.
Genome editing therapy is an experimental approach aimed at correcting faulty genes in order to treat a disease. It can be carried out using several methods, including turning harmful mutations dormant, inserting protective mutations, therapeutic transgenes or distorting viral DNA. The replacement of a mutant allele with a corrected copy of the gene necessitates the successful transport of the latter to an accessible tissue compartment through a carrier, such as a virus or nanoparticle [23].
5. Gene-Editing-Based Therapies
Gene therapy offers a bright prospect for the treatment of numerous illnesses and abnormalities [35]. An experimental treatment known as genome editing therapy tries to fix damaged genes in order to treat AS. Several techniques can be used to implement this new technology, including introducing protective mutations, changing viral DNA, rendering harmful mutations dormant, and using therapeutic transgenes [36].
With good in vitro outcomes in cases of homozygous thalassemia creating functioning red blood cell precursors, the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR/Cas9) system has emerged as a viable gene editing therapy for several uncommon genetic illnesses [36]. According to a recent study by Daga et al. in 2023, it becomes possible to acquire podocyte-lineage cells from urine that reproduce the physiological conditions encountered in these particular cells. This makes it prospective to accurately determine the COL4 variation correction index following experimental interventions [37]. Therefore, employing a self-inactivating dual-plasmid method produced by a self-cleaving streptococcus pyogenes Cas9 (SpCas9), the new technique was employed in AS autosomal dominant variants and X-linked hereditary AS. High correction rates were achieved, ranging from 44% in the COL4A3 gene to 58% in the COL4A5 gene, leading to lower indels (10.4% for COL4A3 and 8.8% for COL4A5) [30]. Despite these encouraging findings, because manipulating podocytes is challenging, there is still a long way to go before this proof of concept can be used within in vivo research.
References
- Williamson, D.A. Alport’s syndrome of hereditary nephritis with deafness. Lancet 1961, 278, 1321–1323.
- Flinter, F. Alport’s syndrome. J. Med. Genet. 1997, 34, 326–330.
- Watson, S.; Padala, S.A.; Hashmi, M.F.; Bush, J.S. Alport Syndrome; StatPearls: Treasure Island, FL, USA, 2017.
- Gibson, J.T.; Huang, M.; Dabrera, M.S.C.; Shukla, K.; Rothe, H.; Hilbert, P.; Deltas, C.; Storey, H.; Lipska-Ziętkiewicz, B.S.; Chan, M.M.Y.; et al. Genotype–phenotype correlations for COL4A3–COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci. Rep. 2022, 12, 2722.
- Gross, O.; Kashtan, C.E. Treatment of Alport syndrome: Beyond animal models. Kidney Int. 2009, 76, 599–603.
- Suh, J.H.; Miner, J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013, 9, 470–477.
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2019, 23, 158–168.
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport syndrome classification and management. Kidney Med. 2020, 2, 639–649.
- Mallett, A.; Tang, W.; Clayton, P.A.; Stevenson, S.; McDonald, S.P.; Hawley, C.M.; Badve, S.V.; Boudville, N.; Brown, F.G.; Campbell, S.B.; et al. End-stage kidney disease due to Alport syndrome: Outcomes in 296 consecutive Australia and New Zealand Dialysis and Transplant Registry cases. Nephrol. Dial. Transplant. 2014, 29, 2277–2286.
- Yamamura, T.; Nozu, K.; Fu, X.J.; Nozu, Y.; Ye, M.J.; Shono, A.; Yamanouchi, S.; Minamikawa, S.; Morisada, N.; Nakanishi, K.; et al. Natural history and genotype–phenotype correlation in female X-linked Alport syndrome. Kidney Int. Rep. 2017, 2, 850–855.
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, O.K.; Flinter, F.; et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657.
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610.
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Faculty Opinions recommendation of Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883.
- Cohen, E.P.; Lemann, J. In hereditary nephritis angiotensin-converting enzyme inhibition decreases proteinuria and may slow the rate of progression. Am. J. Kidney Dis. 1996, 27, 199–203.
- Rheault, M.N.; Savige, J.; Randles, M.J.; Weinstock, A.; Stepney, M.; Turner, A.N.; Parziale, G.; Gross, O.; A Flinter, F.; Miner, J.H.; et al. The importance of clinician, patient and researcher collaborations in Alport syndrome. Pediatr. Nephrol. 2019, 35, 733–742.
- Arrondel, C.; Deschênes, G.; Le Meur, Y.; Viau, A.; Cordonnier, C.; Fournier, A.; Amadeo, S.; Gubler, M.-C.; Antignac, C.; Heidet, L. A large tandem duplication within the COL4A5 gene is responsible for the high prevalence of Alport syndrome in French Polynesia. Kidney Int. 2004, 65, 2030–2040.
- Gale, D.P.; Oygar, D.D.; Lin, F.; Oygar, P.D.; Khan, N.; Connor, T.M.; Lapsley, M.; Maxwell, P.H.; Neild, G.H. A novel COL4A1 frameshift mutation in familial kidney disease: The importance of the C-terminal NC1 domain of type IV collagen. Nephrol. Dial. Transplant. 2016, 31, 1908–1914.
- Savige, J.; Huang, M.; Croos Dabrera, M.S.; Shukla, K.; Gibson, J. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Front. Med. 2022, 9, 865034.
- Savige, J.; Ariani, F.; Mari, F.; Bruttini, M.; Renieri, A.; Gross, O.; Deltas, C.; Flinter, F.; Ding, J.; Gale, D.P.; et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr. Nephrol. 2019, 34, 1175–1189.
- Savige, J.; Storey, H.; Cheong, H.I.; Kang, H.G.; Park, E.; Hilbert, P.; Persikov, A.; Torres-Fernandez, C.; Ars, E.; Torra, R.; et al. X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS ONE 2016, 11, e0161802.
- Savige, J.; Renieri, A.; Ars, E.; Daga, S.; Pinto, A.M.; Rothe, H.; Gale, D.P.; Aksenova, M.; Cerkauskaite, A.; Bielska, O.; et al. Digenic Alport syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 1697–1706.
- Chiereghin, C.; Robusto, M.; Mastrangelo, A.; Castorina, P.; Montini, G.; Giani, M.; Duga, S.; Asselta, R.; Soldà, G. Alport syndrome cold cases: Missing mutations identified by exome sequencing and functional analysis. PLoS ONE 2017, 12, e0178630.
- Chavez, E.; Rodriguez, J.; Drexler, Y.; Fornoni, A. Novel therapies for Alport syndrome. Front. Med. 2022, 9, 848389.
- Bomback, A.S.; Klemmer, P.J. The incidence and implications of aldosterone breakthrough. Nat. Clin. Pract. Nephrol. 2007, 3, 486–492.
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446.
- Liu, J.; Cui, J.; Fang, X.; Chen, J.; Yan, W.; Shen, Q.; Xu, H. Efficacy and safety of dapagliflozin in children with inherited proteinuric kidney disease: A pilot study. Kidney Int. Rep. 2022, 7, 638–641.
- Palmer, S.C.; Strippoli, G.F.; Craig, J.C. KHA-CARI commentary on the KDIGO clinical practice guideline for lipid management in chronic kidney disease. Nephrology 2014, 19, 663–666.
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of different types of statins on kidney function decline and proteinuria: A network meta-analysis. Sci. Rep. 2019, 9, 16632.
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68.
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156.
- Gross, O.; Koepke, M.L.; Beirowski, B.; Schulze-Lohoff, E.; Segerer, S.; Weber, M. Nephroprotection by antifibrotic and anti-inflammatory effects of the vasopeptidase inhibitor AVE7688. Kidney Int. 2005, 68, 456–463.
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Müller, G.A.; Kalluri, R.; Takayanagi, K.; Shimizu, T.; Tayama, Y.; et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003, 285, F1060–F1067.
- Ninichuk, V.; Gross, O.; Reichel, C.; Khandoga, A.; Pawar, R.D.; Ciubar, R.; Segerer, S.; Belemezova, E.; Radomska, E.; Luckow, B.; et al. Anders HJ. Faculty Opinions recommendation of Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J. Am. Soc. Nephrol. 2005, 16, 977–985.
- Sugimoto, H.; Mundel, T.M.; Sund, M.; Xie, L.; Cosgrove, D.; Kalluri, R. Faculty Opinions recommendation of Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 7321–7326.
- Hawsawi, Y.M.; Shams, A.; Theyab, A.; Siddiqui, J.; Barnawee, M.; Abdali, W.A.; Marghalani, N.A.; Alshelali, N.H.; Al-Sayed, R.; Alzahrani, O.; et al. The State-of-the-Art of Gene Editing and its Application to Viral Infections and Diseases Including COVID-19. Front. Cell. Infect. Microbiol. 2022, 12, 869889.
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131.
- Daga, S.; Donati, F.; Capitani, K.; Croci, S.; Tita, R.; Giliberti, A.; Valentino, F.; Benetti, E.; Fallerini, C.; Niccheri, F.; et al. Correction: New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 2020, 28, 480–490, Erratum in Eur. J. Hum. Genet. 2023, Online ahead of print.
More
Information
Subjects:
Clinical Neurology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
786
Revisions:
3 times
(View History)
Update Date:
23 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No