Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yousef Hawsawi and Version 3 by Lindsay Dong.
Alport syndrome (AS) is a rare genetic disorder categorized by the progressive loss of kidney function, sensorineural hearing loss and eye abnormalities. It occurs due to mutations in three genes that encode for the alpha chains of type IV collagen. Globally, the disease is classified based on the pattern of inheritance into X-linked AS (XLAS), which is caused by pathogenic variants in COL4A5, representing 80% of AS. Autosomal recessive AS (ARAS), caused by mutations in either COL4A3 or COL4A4, represents 15% of AS. Autosomal dominant AS (ADAS) is rare and has been recorded in 5% of all cases due to mutations in COL4A3 or COL4A4.
- Alport syndrome
- type IV collagen
- glomerular basement membrane
- kidney disease
- gene technology
1. Introduction
In 1875, Dickinson was the first scientist who reported the presence of familial inherited renal failure. In 1972, Arthur Cecil Alport was the first British physician who described symptoms of kidney and hearing health problems in families. He pointed out that females were less severely affected by deafness than males, although the disorder was more likely transmitted by females. Further cases were diagnosed, and SAS was approved in 1961 as an eponym [1][2][1,2]. It has been observed that AS develops frequently in patients with hematuria (i.e., bloody urine), nerve deafness, edema, and hypertension [3].
Alport syndrome is significantly caused by mutations in one of three genes that code for type IV collagen alpha chains: COL4A3 (α3), COL4A4 (α4) and/or COL4A5 (α5) [4]. Those genes are involved in type IV collagen biosynthesis [5]. Type IV collagen mainly constitutes of the glomerular basement membrane (GBM), which represents ~50% of its total protein mass and is responsible for its stability. However, GBM is a thin extracellular matrix protein that functions as a per selective barrier to the passage of blood cells and proteins from the blood to the urinary tract [6] (Figure 1(3)). Changes in α-chains can give rise to dysfunctional GBM, leading to sensorineural deafness, abnormalities in several parts of the eyes, hematuria, proteinuria (i.e., excessive protein in urine) and eventual chronic kidney disease (CKD) [7][8][7,8].
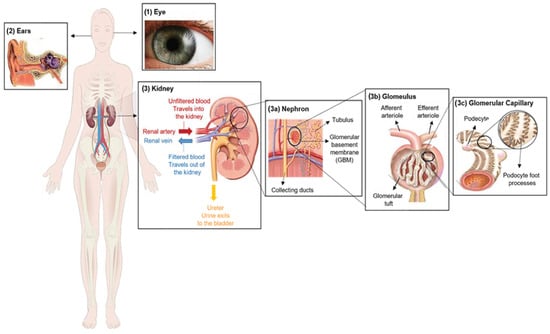
Figure 1. Clinical characteristics of Alport syndrome in individuals. Symptoms of Alport syndrome can be categorized by (1) eye abnormalities; (2) sensorineural hearing loss; and (3) related kidney symptoms.
Estimations of gene mutation frequency range from 1 in 5000 to 1 in 10,000, demonstrating its high burden in clinical settings [9]. As a rare disease, the estimated frequency of AS is approximately 1 in 50,000 live births worldwide [3]. Despite advances in therapeutic strategies that are performed to address many of the associated symptoms and slow the progression of kidney disease, there is no radical therapy for AS as of this moment.
Alport syndrome (AS) is a rare genetic disorder categorized by a progressive loss of kidney function, sensorineural hearing loss and eye abnormalities (Figure 1).
2. Clinical Characterization of Alport Syndrome
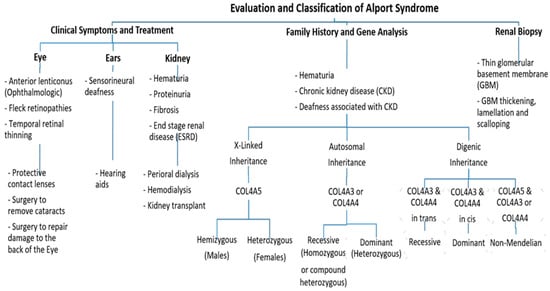
Clinically, AS characteristics are diverse, with a wide range progressing to end-stage renal disease (ESRD). The main and initial observed criterion among all the AS modes of inheritance is persistent hematuria, as approximately detected in all male and 97% of female patients [10]. Proteinuria is detected in male patients in early childhood, with levels gradually increasing with age, and occasionally presents as a nephrotic condition. It is also estimated that 90% of Proteinuric patients acquire ESRD by the age of 40 years, with the median age of ESRD development being 25 years. By the age of 40, 12% of females have acquired ESRD [10][11][10,11]. Also, Nozu et al. in 2019 reported that female XLAS patients developed proteinuria at a median age of 7 years and ESRD at a median age of 65 years [7]. Ultimately, 90% of male and 20% of female patients develop end-stage renal disease (ESRD) by the age of 40 years [11][12][11,12]. Further, ophthalmologic consequences include specific ocular abnormalities such as anterior lenticonus, posterior subcapsular cataract, posterior polymorphous dystrophy, and retinal flecks [13][14][13,14]. In addition, AS-affected patients frequently exhibit external disorders such as sensorineural deafness, of which frequently develops in late infancy; by the age of 40, 90% of male patients and around 12% of female patients have hearing loss [11][12][11,12] (Figure 2).Figure 2. An approach regarding the evaluation and classification of Alport syndrome in individuals. In X-Linked Alport syndrome (X-Linked AS), the disease is usually passed from the mother to her child. In autosomal recessive Alport syndrome (ARAS), both sides of the family must have the mutation for AS to be passed on to their children, while in autosomal dominant Alport syndrome (ADAS), only one side of the family must have the mutation to pass it.
3. Genetics of Alport Syndrome
There are six members of type IV collagen (COL4A1, COL4A2, COL4A3, COL4A4, COL4A5 and COL4A6), which encode six unique alpha chains of type IV collagen (α1–α6) (Figure 3).
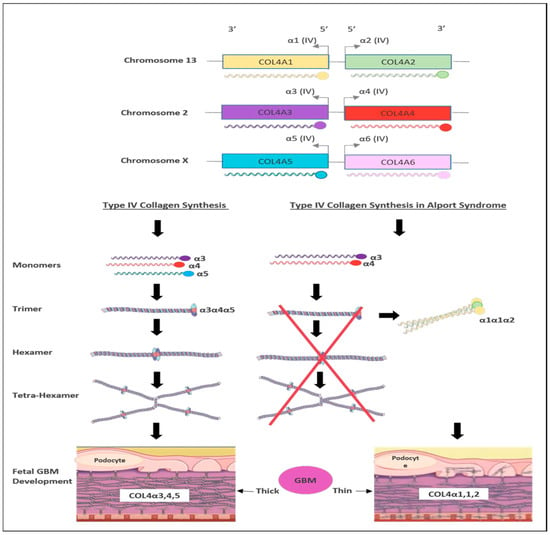
Figure 3. Schematic representation of type IV collagen biosynthesis. Type IV collagen genes are located in three different chromosomes pairwise, which encode six unique α-chains of type IV collagen (α1–α6). Monomers of collagen α3α4α5 can be combined among other forming triple helices (trimers). Two trimers then combine to form hexamers that can be associated with additional three hexamers that form tera-hexamer, which forms a thick GBM layer. However, in the absence of one monomer of the three α-chains, the kidney produces a monomer composed of α1α1α2 that eventually creates a thin GBM layer.
Pathogenic variants in these genes can cause several progressive and non-progressive glomerular disorders [15][21]. Mutations in COL4A5 are the main cause of XLAS (Figure 2) and are responsible for more than 80% of AS cases [4]. It is located on chromosome Xq22 and has approximately 51 exons that encode 1685 amino acids [16][22]. The protein structure of the α5 chain includes three parts: A signal peptide with 26 amino acid residues, a collagenous domain with 1430 residues that consist of short non-collagenous interruptions and a 229 amino acid carboxy-terminal non-collagenous (NC) domain [11].
On the other hand, mutations in COL4A3 or COL4A4 genes are the main cause of autosomal recessive AS (ARAS) or autosomal dominant AS (ADAS) (Figure 2). Those genes are arranged head-to-head on chromosome 2 (2q36.3) and both share one promotor that encodes other type IV collagen genes [17][26]. The COL4A3 gene consists of 52 exons that span over 88 kb and encode 1670 amino acids, whereas COL4A4 spans 161 kb and is composed of 48 exons [4]. Both COL4A3 and COL4A4 proteins are composed of the N-terminal amino acid domain, the central triple-helical domain including G-X-Y repeats, and the C-terminal globular non-collagenous domain with a highly homologous protein structure with COL4A5 [18][27].
The ARAS mutations in either COL4A3 or COL4A4 are responsible for 15% of AS patients with no family history of AS [19][28]. Individuals with ARAS exhibit clinical features that are similar to those seen in males with XLAS. Carriers with the monoallelic variant are frequently asymptomatic or show only mild proteinuria and microscopic hematuria [20][24]. However, individuals with truncating variants are more likely to develop kidney failure before the age of 30 [19][21][28,29].
The genotype–phenotype correlation has been studied in a European cohort [11]. Earlier genotype–phenotype correlation studies have revealed that males with large deletions, frameshift mutations, and truncating variants in the COL4A5 gene present with more severe clinical manifestations and are at a high risk of kidney failure at a younger age [11], whereas males with missense variants tend to exhibit milder disease features. In contrast, no clear genotype–phenotype correlation has been reported among females with heterozygous variants in COL4A5 and their age at kidney failure [12]. However, it has been observed that females with missense variants in the COL4A5 gene have better kidney function and are less likely to develop proteinuria in comparison to females carrying other types of variants [22][35]. A study by Bekheirnia et al. from 2010 examined the relationships between genotype and phenotype in a sizable US cohort of male patients with XLAS. As an outcome, they discovered that missense mutations (51% of the families) were most frequently found, followed by truncating (14%) and splice site (13.7%) alterations [13].
The most common pathogenic variants are the missense variants that affect Gly resides in the collagenous Gly Xaa Yaa repeats, which cause AS in Gly substitutions [4]. The clinical features and severity associated with Gly missense variants highly vary between patients [18][27]. In contrast, the mutation substitutions affecting non-collagenous boundary Gly residues resulted in kidney failure at a delayed age [4].