Alport syndrome (AS) is a rare genetic disorder categorized by the progressive loss of kidney function, sensorineural hearing loss and eye abnormalities. It occurs due to mutations in three genes that encode for the alpha chains of type IV collagen. Globally, the disease is classified based on the pattern of inheritance into X-linked AS (XLAS), which is caused by pathogenic variants in COL4A5, representing 80% of AS. Autosomal recessive AS (ARAS), caused by mutations in either COL4A3 or COL4A4, represents 15% of AS. Autosomal dominant AS (ADAS) is rare and has been recorded in 5% of all cases due to mutations in COL4A3 or COL4A4.
- Alport syndrome
- type IV collagen
- glomerular basement membrane
- kidney disease
- gene technology
1. Introduction
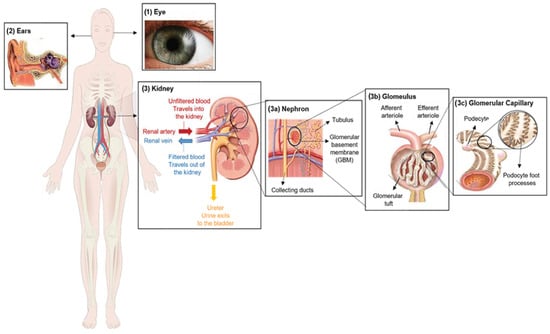
2. Clinical Characterization of Alport Syndrome
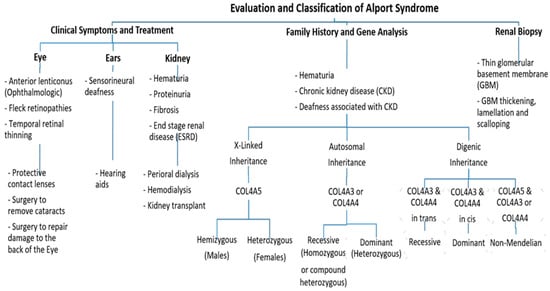
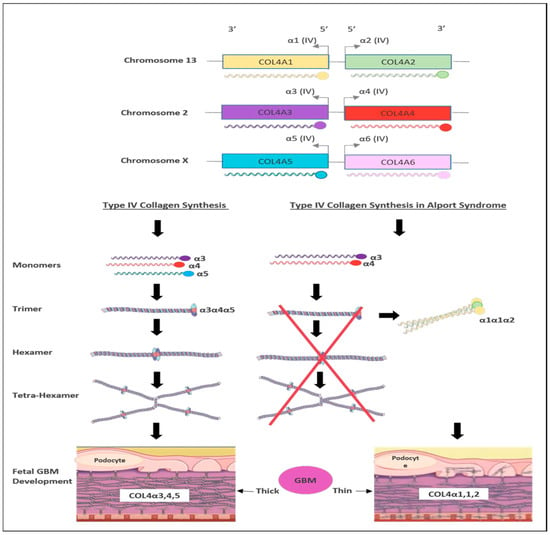
3. Genetics of Alport Syndrome
4. Current Therapies for Alport Syndrome Management
4.1. Chemical Drugs
4.2. Molecular Therapies
5. Gene-Editing-Based Therapies
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11102762
References
- Williamson, D.A. Alport’s syndrome of hereditary nephritis with deafness. Lancet 1961, 278, 1321–1323.
- Flinter, F. Alport’s syndrome. J. Med. Genet. 1997, 34, 326–330.
- Watson, S.; Padala, S.A.; Hashmi, M.F.; Bush, J.S. Alport Syndrome; StatPearls: Treasure Island, FL, USA, 2017.
- Gibson, J.T.; Huang, M.; Dabrera, M.S.C.; Shukla, K.; Rothe, H.; Hilbert, P.; Deltas, C.; Storey, H.; Lipska-Ziętkiewicz, B.S.; Chan, M.M.Y.; et al. Genotype–phenotype correlations for COL4A3–COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci. Rep. 2022, 12, 2722.
- Gross, O.; Kashtan, C.E. Treatment of Alport syndrome: Beyond animal models. Kidney Int. 2009, 76, 599–603.
- Suh, J.H.; Miner, J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013, 9, 470–477.
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2019, 23, 158–168.
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport syndrome classification and management. Kidney Med. 2020, 2, 639–649.
- Mallett, A.; Tang, W.; Clayton, P.A.; Stevenson, S.; McDonald, S.P.; Hawley, C.M.; Badve, S.V.; Boudville, N.; Brown, F.G.; Campbell, S.B.; et al. End-stage kidney disease due to Alport syndrome: Outcomes in 296 consecutive Australia and New Zealand Dialysis and Transplant Registry cases. Nephrol. Dial. Transplant. 2014, 29, 2277–2286.
- Yamamura, T.; Nozu, K.; Fu, X.J.; Nozu, Y.; Ye, M.J.; Shono, A.; Yamanouchi, S.; Minamikawa, S.; Morisada, N.; Nakanishi, K.; et al. Natural history and genotype–phenotype correlation in female X-linked Alport syndrome. Kidney Int. Rep. 2017, 2, 850–855.
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, O.K.; Flinter, F.; et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657.
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610.
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Faculty Opinions recommendation of Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883.
- Cohen, E.P.; Lemann, J. In hereditary nephritis angiotensin-converting enzyme inhibition decreases proteinuria and may slow the rate of progression. Am. J. Kidney Dis. 1996, 27, 199–203.
- Rheault, M.N.; Savige, J.; Randles, M.J.; Weinstock, A.; Stepney, M.; Turner, A.N.; Parziale, G.; Gross, O.; A Flinter, F.; Miner, J.H.; et al. The importance of clinician, patient and researcher collaborations in Alport syndrome. Pediatr. Nephrol. 2019, 35, 733–742.
- Arrondel, C.; Deschênes, G.; Le Meur, Y.; Viau, A.; Cordonnier, C.; Fournier, A.; Amadeo, S.; Gubler, M.-C.; Antignac, C.; Heidet, L. A large tandem duplication within the COL4A5 gene is responsible for the high prevalence of Alport syndrome in French Polynesia. Kidney Int. 2004, 65, 2030–2040.
- Gale, D.P.; Oygar, D.D.; Lin, F.; Oygar, P.D.; Khan, N.; Connor, T.M.; Lapsley, M.; Maxwell, P.H.; Neild, G.H. A novel COL4A1 frameshift mutation in familial kidney disease: The importance of the C-terminal NC1 domain of type IV collagen. Nephrol. Dial. Transplant. 2016, 31, 1908–1914.
- Savige, J.; Huang, M.; Croos Dabrera, M.S.; Shukla, K.; Gibson, J. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Front. Med. 2022, 9, 865034.
- Savige, J.; Ariani, F.; Mari, F.; Bruttini, M.; Renieri, A.; Gross, O.; Deltas, C.; Flinter, F.; Ding, J.; Gale, D.P.; et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr. Nephrol. 2019, 34, 1175–1189.
- Savige, J.; Storey, H.; Cheong, H.I.; Kang, H.G.; Park, E.; Hilbert, P.; Persikov, A.; Torres-Fernandez, C.; Ars, E.; Torra, R.; et al. X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS ONE 2016, 11, e0161802.
- Savige, J.; Renieri, A.; Ars, E.; Daga, S.; Pinto, A.M.; Rothe, H.; Gale, D.P.; Aksenova, M.; Cerkauskaite, A.; Bielska, O.; et al. Digenic Alport syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 1697–1706.
- Chiereghin, C.; Robusto, M.; Mastrangelo, A.; Castorina, P.; Montini, G.; Giani, M.; Duga, S.; Asselta, R.; Soldà, G. Alport syndrome cold cases: Missing mutations identified by exome sequencing and functional analysis. PLoS ONE 2017, 12, e0178630.
- Chavez, E.; Rodriguez, J.; Drexler, Y.; Fornoni, A. Novel therapies for Alport syndrome. Front. Med. 2022, 9, 848389.
- Bomback, A.S.; Klemmer, P.J. The incidence and implications of aldosterone breakthrough. Nat. Clin. Pract. Nephrol. 2007, 3, 486–492.
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446.
- Liu, J.; Cui, J.; Fang, X.; Chen, J.; Yan, W.; Shen, Q.; Xu, H. Efficacy and safety of dapagliflozin in children with inherited proteinuric kidney disease: A pilot study. Kidney Int. Rep. 2022, 7, 638–641.
- Palmer, S.C.; Strippoli, G.F.; Craig, J.C. KHA-CARI commentary on the KDIGO clinical practice guideline for lipid management in chronic kidney disease. Nephrology 2014, 19, 663–666.
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of different types of statins on kidney function decline and proteinuria: A network meta-analysis. Sci. Rep. 2019, 9, 16632.
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68.
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156.
- Gross, O.; Koepke, M.L.; Beirowski, B.; Schulze-Lohoff, E.; Segerer, S.; Weber, M. Nephroprotection by antifibrotic and anti-inflammatory effects of the vasopeptidase inhibitor AVE7688. Kidney Int. 2005, 68, 456–463.
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Müller, G.A.; Kalluri, R.; Takayanagi, K.; Shimizu, T.; Tayama, Y.; et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003, 285, F1060–F1067.
- Ninichuk, V.; Gross, O.; Reichel, C.; Khandoga, A.; Pawar, R.D.; Ciubar, R.; Segerer, S.; Belemezova, E.; Radomska, E.; Luckow, B.; et al. Anders HJ. Faculty Opinions recommendation of Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J. Am. Soc. Nephrol. 2005, 16, 977–985.
- Sugimoto, H.; Mundel, T.M.; Sund, M.; Xie, L.; Cosgrove, D.; Kalluri, R. Faculty Opinions recommendation of Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 7321–7326.
- Hawsawi, Y.M.; Shams, A.; Theyab, A.; Siddiqui, J.; Barnawee, M.; Abdali, W.A.; Marghalani, N.A.; Alshelali, N.H.; Al-Sayed, R.; Alzahrani, O.; et al. The State-of-the-Art of Gene Editing and its Application to Viral Infections and Diseases Including COVID-19. Front. Cell. Infect. Microbiol. 2022, 12, 869889.
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131.
- Daga, S.; Donati, F.; Capitani, K.; Croci, S.; Tita, R.; Giliberti, A.; Valentino, F.; Benetti, E.; Fallerini, C.; Niccheri, F.; et al. Correction: New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 2020, 28, 480–490, Erratum in Eur. J. Hum. Genet. 2023, Online ahead of print.