 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gjin Ndrepepa | -- | 6347 | 2023-08-31 20:32:02 | | | |
| 2 | Dean Liu | Meta information modification | 6347 | 2023-09-01 03:23:36 | | |
Video Upload Options
Coronary no-reflow (CNR) is a frequent phenomenon that develops in patients with ST-segment elevation myocardial infarction (STEMI) following reperfusion therapy. CNR is highly dynamic, develops gradually (over hours) and persists for days to weeks after reperfusion. Microvascular obstruction (MVO) developing as a consequence of myocardial ischemia, distal embolization and reperfusion-related injury is the main pathophysiological mechanism of CNR. The frequency of CNR or MVO after primary PCI differs widely depending on the sensitivity of the tools used for diagnosis and timing of examination. Coronary angiography is readily available and most convenient to diagnose CNR but it is highly conservative and underestimates the true frequency of CNR. Cardiac magnetic resonance (CMR) imaging is the most sensitive method to diagnose MVO and CNR that provides information on the presence, localization and extent of MVO. CMR imaging detects intramyocardial hemorrhage and accurately estimates the infarct size. MVO and CNR markedly negate the benefits of reperfusion therapy and contribute to poor clinical outcomes including adverse remodeling of left ventricle, worsening or new congestive heart failure and reduced survival.
1. Historical Perspective
2. Pathophysiology of CNR
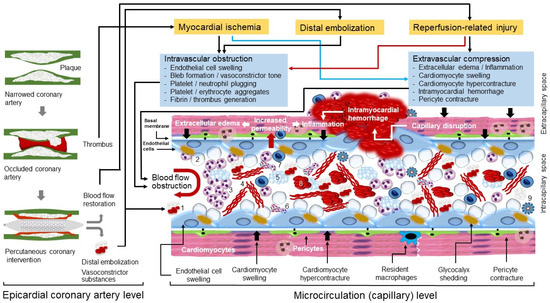
2.1. A Short Description of Myocardial Microcirculation
2.2. Myocardial Ischemia
2.3. Distal Embolization
2.4. Reperfusion-Related Injury
References
- Majno, G.; Ames, A.; Chiang, J.; Wright, R.L. No Reflow after Cerebral Ischaemia. Lancet 1967, 2, 569–570.
- Ames, A., 3rd; Wright, R.L.; Kowada, M.; Thurston, J.M.; Majno, G. Cerebral ischemia. II. The no-reflow phenomenon. Am. J. Pathol. 1968, 52, 437–453.
- Chiang, J.; Kowada, M.; Ames, A., 3rd; Wright, R.L.; Majno, G. Cerebral ischemia. III. Vascular changes. Am. J. Pathol. 1968, 52, 455–476.
- Harman, J.W. The Significance of Local Vascular Phenomena in the Production of Ischemic Necrosis in Skeletal Muscle. Am. J. Pathol. 1948, 24, 625–641.
- Sheehan, H.L.; Davis, J.C. Renal Ischaemia with Failed Reflow. J. Pathol. Bacteriol. 1959, 78, 105–120.
- Flores, J.; DiBona, D.R.; Beck, C.H.; Leaf, A. The role of cell swelling in ischemic renal damage and the protective effect of hypertonic solute. J. Clin. Investig. 1972, 51, 118–126.
- Kovacs, K.; Carroll, R.; Tapp, E. Temporary Ischaemia of Rat Adrenal Gland. J. Pathol. Bacteriol. 1966, 91, 235–240.
- Krug, A.; De Rochemont, W.D.M.; Korb, G. Blood supply of the myocardium after temporary coronary occlusion. Circ. Res. 1966, 19, 57–62.
- Willms-Kretschmer, K.; Majno, G. Ischemia of the skin. Electron microscopic study of vascular injury. Am. J. Pathol. 1969, 54, 327–353.
- Kloner, R.A.; Ganote, C.E.; Jennings, R.B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Investig. 1974, 54, 1496–1508.
- Kloner, R.A.; Ganote, C.E.; Whalen, D.A., Jr.; Jennings, R.B. Effect of a transient period of ischemia on myocardial cells. II. Fine structure during the first few minutes of reflow. Am. J. Pathol. 1974, 74, 399–422.
- Jefferson, G. “Arterial Embolectomy”. Br. Med. J. 1934, 2, 1090–1094.
- Griffiths, D.L. Volkmann’s ischaemic contracture. Br. J. Surg. 1940, 28, 239–260.
- Schofer, J.; Montz, R.; Mathey, D.G. Scintigraphic evidence of the “no reflow” phenomenon in human beings after coronary thrombolysis. J. Am. Coll. Cardiol. 1985, 5, 593–598.
- Bates, E.R.; Krell, M.J.; Dean, E.N.; O’Neill, W.W.; Vogel, R.A. Demonstration of the “no-reflow” phenomenon by digital coronary arteriography. Am. J. Cardiol. 1986, 57, 177–178.
- Pomerantz, R.M.; Kuntz, R.E.; Diver, D.J.; Safian, R.D.; Baim, D.S. Intracoronary verapamil for the treatment of distal microvascular coronary artery spasm following PTCA. Cathet Cardiovasc. Diagn. 1991, 24, 283–285.
- Feld, H.; Lichstein, E.; Schachter, J.; Shani, J. Early and late angiographic findings of the “no-reflow” phenomenon following direct angioplasty as primary treatment for acute myocardial infarction. Am. Heart J. 1992, 123, 782–784.
- Wilson, R.F.; Laxson, D.D.; Lesser, J.R.; White, C.W. Intense microvascular constriction after angioplasty of acute thrombotic coronary arterial lesions. Lancet 1989, 1, 807–811.
- Ito, H.; Tomooka, T.; Sakai, N.; Yu, H.; Higashino, Y.; Fujii, K.; Masuyama, T.; Kitabatake, A.; Minamino, T. Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation 1992, 85, 1699–1705.
- Piana, R.N.; Paik, G.Y.; Moscucci, M.; Cohen, D.J.; Gibson, C.M.; Kugelmass, A.D.; Carrozza, J.P., Jr.; Kuntz, R.E.; Baim, D.S. Incidence and treatment of ‘no-reflow’ after percutaneous coronary intervention. Circulation 1994, 89, 2514–2518.
- Morishima, I.; Sone, T.; Mokuno, S.; Taga, S.; Shimauchi, A.; Oki, Y.; Kondo, J.; Tsuboi, H.; Sassa, H. Clinical significance of no-reflow phenomenon observed on angiography after successful treatment of acute myocardial infarction with percutaneous transluminal coronary angioplasty. Am. Heart J. 1995, 130, 239–243.
- Morishima, I.; Sone, T.; Okumura, K.; Tsuboi, H.; Kondo, J.; Mukawa, H.; Matsui, H.; Toki, Y.; Ito, T.; Hayakawa, T. Angiographic no-reflow phenomenon as a predictor of adverse long-term outcome in patients treated with percutaneous transluminal coronary angioplasty for first acute myocardial infarction. J. Am. Coll. Cardiol. 2000, 36, 1202–1209.
- Rakusan, K.; Flanagan, M.F.; Geva, T.; Southern, J.; Van Praagh, R. Morphometry of human coronary capillaries during normal growth and the effect of age in left ventricular pressure-overload hypertrophy. Circulation 1992, 86, 38–46.
- Kaul, S.; Jayaweera, A.R. Coronary and myocardial blood volumes: Noninvasive tools to assess the coronary microcirculation? Circulation 1997, 96, 719–724.
- Kaul, S.; Ito, H. Microvasculature in acute myocardial ischemia: Part I: Evolving concepts in pathophysiology, diagnosis, and treatment. Circulation 2004, 109, 146–149.
- Kurose, I.; Anderson, D.C.; Miyasaka, M.; Tamatani, T.; Paulson, J.C.; Todd, R.F.; Rusche, J.R.; Granger, D.N. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ. Res. 1994, 74, 336–343.
- Heusch, G. The Coronary Circulation as a Target of Cardioprotection. Circ. Res. 2016, 118, 1643–1658.
- Stempien-Otero, A.; Karsan, A.; Cornejo, C.J.; Xiang, H.; Eunson, T.; Morrison, R.S.; Kay, M.; Winn, R.; Harlan, J. Mechanisms of hypoxia-induced endothelial cell death. Role of p53 in apoptosis. J. Biol. Chem. 1999, 274, 8039–8045.
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170.
- Baldea, I.; Teacoe, I.; Olteanu, D.E.; Vaida-Voievod, C.; Clichici, A.; Sirbu, A.; Filip, G.A.; Clichici, S. Effects of different hypoxia degrees on endothelial cell cultures-Time course study. Mech. Ageing Dev. 2018, 172, 45–50.
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246.
- Steenbergen, C.; Hill, M.L.; Jennings, R.B. Volume regulation and plasma membrane injury in aerobic, anaerobic, and ischemic myocardium in vitro. Effects of osmotic cell swelling on plasma membrane integrity. Circ. Res. 1985, 57, 864–875.
- Kasseckert, S.A.; Schafer, C.; Kluger, A.; Gligorievski, D.; Tillmann, J.; Schluter, K.D.; Noll, T.; Sauer, H.; Piper, H.M.; Abdallah, Y. Stimulation of cGMP signalling protects coronary endothelium against reperfusion-induced intercellular gap formation. Cardiovasc. Res. 2009, 83, 381–387.
- Patterson, C.E.; Lum, H. Update on pulmonary edema: The role and regulation of endothelial barrier function. Endothelium 2001, 8, 75–105.
- Waschke, J.; Curry, F.E.; Adamson, R.H.; Drenckhahn, D. Regulation of actin dynamics is critical for endothelial barrier functions. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1296–H1305.
- Ndrepepa, G.; Colleran, R.; Kastrati, A. Reperfusion injury in ST-segment elevation myocardial infarction: The final frontier. Coron. Artery Dis. 2017, 28, 253–262.
- Maxwell, L.; Gavin, J.B. The role of post-ischaemic reperfusion in the development of microvascular incompetence and ultrastructural damage in the myocardium. Basic Res. Cardiol. 1991, 86, 544–553.
- Dvorak, H.F. Discovery of vascular permeability factor (VPF). Exp. Cell Res. 2006, 312, 522–526.
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504.
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1208.
- Bouleti, C.; Mewton, N.; Germain, S. The no-reflow phenomenon: State of the art. Arch. Cardiovasc. Dis. 2015, 108, 661–674.
- Feng, Y.; Venema, V.J.; Venema, R.C.; Tsai, N.; Behzadian, M.A.; Caldwell, R.B. VEGF-induced permeability increase is mediated by caveolae. Investig. Ophthalmol. Vis. Sci. 1999, 40, 157–167.
- Scotland, R.S.; Cohen, M.; Foster, P.; Lovell, M.; Mathur, A.; Ahluwalia, A.; Hobbs, A.J. C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc. Natl. Acad. Sci. USA 2005, 102, 14452–14457.
- Kerner, T.; Ahlers, O.; Reschreiter, H.; Buhrer, C.; Mockel, M.; Gerlach, H. Adhesion molecules in different treatments of acute myocardial infarction. Crit. Care 2001, 5, 145–150.
- Vink, H.; Duling, B.R. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ. Res. 1996, 79, 581–589.
- Kolarova, H.; Ambruzova, B.; Sindlerova, L.S.; Klinke, A.; Kubala, L. Modulation of Endothelial Glycocalyx Structure under Inflammatory Conditions. Mediat. Inflamm. 2014, 2014, 694312.
- Ostergaard, L.; Kristiansen, S.B.; Angleys, H.; Frokiaer, J.; Hasenkam, J.M.; Jespersen, S.N.; Botker, H.E. The role of capillary transit time heterogeneity in myocardial oxygenation and ischemic heart disease. Basic Res. Cardiol. 2014, 109, 409.
- Ishiharajima, S.; Aida, T.; Nakagawa, R.; Kameyama, K.; Sugano, K.; Oguro, T.; Asano, G. Early membrane damage during ischemia in rat heart. Exp. Mol. Pathol. 1986, 44, 1–6.
- Czarnowska, E.; Karwatowskaprokopczuk, E. Ultrastructural Demonstration of Endothelial Glycocalyx Disruption in the Reperfused Rat-Heart—Involvement of Oxygen-Free Radicals. Basic Res. Cardiol. 1995, 90, 357–364.
- Rubio-Gayosso, I.; Platts, S.H.; Duling, B.R. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2247–H2256.
- Vink, H.; Constantinescu, A.A.; Spaan, J.A. Oxidized lipoproteins degrade the endothelial surface layer: Implications for platelet-endothelial cell adhesion. Circulation 2000, 101, 1500–1502.
- Nieuwdorp, M.; Mooij, H.L.; Kroon, J.; Atasever, B.; Spaan, J.A.; Ince, C.; Holleman, F.; Diamant, M.; Heine, R.J.; Hoekstra, J.B.; et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 2006, 55, 1127–1132.
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89.
- Granger, D.N.; Kvietys, P.R. Reperfusion therapy-What’s with the obstructed, leaky and broken capillaries? Pathophysiology 2017, 24, 213–228.
- Hahn, R.G.; Patel, V.; Dull, R.O. Human glycocalyx shedding: Systematic review and critical appraisal. Acta Anaesthesiol. Scand. 2021, 65, 590–606.
- Bruegger, D.; Rehm, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Conzen, P.; Becker, B.F. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit. Care 2008, 12, R73.
- van den Berg, B.M.; Vink, H.; Spaan, J.A. The endothelial glycocalyx protects against myocardial edema. Circ. Res. 2003, 92, 592–594.
- Chappell, D.; Dorfler, N.; Jacob, M.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 2010, 34, 133–139.
- Chappell, D.; Brettner, F.; Doerfler, N.; Jacob, M.; Rehm, M.; Bruegger, D.; Conzen, P.; Jacob, B.; Becker, B.F. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion: An animal study. Eur. J. Anaesthesiol. 2014, 31, 474–481.
- Bonaventura, A.; Montecucco, F.; Dallegri, F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur. J. Clin. Investig. 2016, 46, 590–601.
- Charron, T.; Jaffe, R.; Segev, A.; Bang, K.W.; Qiang, B.; Sparkes, J.D.; Butany, J.; Dick, A.J.; Freedman, J.; Strauss, B.H. Effects of distal embolization on the timing of platelet and inflammatory cell activation in interventional coronary no-reflow. Thromb. Res. 2010, 126, 50–55.
- Konijnenberg, L.S.F.; Damman, P.; Duncker, D.J.; Kloner, R.A.; Nijveldt, R.; van Geuns, R.M.; Berry, C.; Riksen, N.P.; Escaned, J.; van Royen, N. Pathophysiology and diagnosis of coronary microvascular dysfunction in ST-elevation myocardial infarction. Cardiovasc. Res. 2020, 116, 787–805.
- Battinelli, E.M.; Markens, B.A.; Italiano, J.E., Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood 2011, 118, 1359–1369.
- Ziegler, M.; Wang, X.; Peter, K. Platelets in cardiac ischaemia/reperfusion injury: A promising therapeutic target. Cardiovasc. Res. 2019, 115, 1178–1188.
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G.; Action, E.-C.C. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155.
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front. Immunol. 2019, 10, 1260.
- Yang, B.C.; Virmani, R.; Nichols, W.W.; Mehta, J.L. Platelets protect against myocardial dysfunction and injury induced by ischemia and reperfusion in isolated rat hearts. Circ. Res. 1993, 72, 1181–1190.
- Heindl, B.; Zahler, S.; Welsch, U.; Becker, B.F. Disparate effects of adhesion and degranulation of platelets on myocardial and coronary function in postischaemic hearts. Cardiovasc. Res. 1998, 38, 383–394.
- Choudhri, T.F.; Hoh, B.L.; Zerwes, H.G.; Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S., Jr.; Kottirsch, G.; Pinsky, D.J. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J. Clin. Investig. 1998, 102, 1301–1310.
- Golino, P.; Maroko, P.R.; Carew, T.E. Efficacy of platelet depletion in counteracting the detrimental effect of acute hypercholesterolemia on infarct size and the no-reflow phenomenon in rabbits undergoing coronary artery occlusion-reperfusion. Circulation 1987, 76, 173–180.
- Neumann, F.J.; Blasini, R.; Schmitt, C.; Alt, E.; Dirschinger, J.; Gawaz, M.; Kastrati, A.; Schomig, A. Effect of glycoprotein IIb/IIIa receptor blockade on recovery of coronary flow and left ventricular function after the placement of coronary-artery stents in acute myocardial infarction. Circulation 1998, 98, 2695–2701.
- Ong, S.B.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87.
- Gotz, A.K.; Zahler, S.; Stumpf, P.; Welsch, U.; Becker, B.F. Intracoronary formation and retention of micro aggregates of leukocytes and platelets contribute to postischemic myocardial dysfunction. Basic Res. Cardiol. 2005, 100, 413–421.
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509.
- Jaeschke, H.; Smith, C.W. Mechanisms of neutrophil-induced parenchymal cell injury. J. Leukoc. Biol. 1997, 61, 647–653.
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51.
- Romanic, A.M.; Harrison, S.M.; Bao, W.; Burns-Kurtis, C.L.; Pickering, S.; Gu, J.; Grau, E.; Mao, J.; Sathe, G.M.; Ohlstein, E.H.; et al. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardiovasc. Res. 2002, 54, 549–558.
- Lindsey, M.; Wedin, K.; Brown, M.D.; Keller, C.; Evans, A.J.; Smolen, J.; Burns, A.R.; Rossen, R.D.; Michael, L.; Entman, M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001, 103, 2181–2187.
- Nees, S.; Weiss, D.R.; Senftl, A.; Knott, M.; Forch, S.; Schnurr, M.; Weyrich, P.; Juchem, G. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: The second most frequent myocardial cell type in vitro. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H69–H84.
- O’Farrell, F.M.; Mastitskaya, S.; Hammond-Haley, M.; Freitas, F.; Wah, W.R.; Attwell, D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife 2017, 6, e29280.
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60.
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037.
- Costa, M.A.; Paiva, A.E.; Andreotti, J.P.; Cardoso, M.V.; Cardoso, C.D.; Mintz, A.; Birbrair, A. Pericytes constrict blood vessels after myocardial ischemia. J. Mol. Cell Cardiol. 2018, 116, 1–4.
- Methner, C.; Cao, Z.; Mishra, A.; Kaul, S. Mechanism and potential treatment of the “no reflow” phenomenon after acute myocardial infarction: Role of pericytes and GPR39. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H1030–H1041.
- Li, Q.; Guo, Z.; Wu, C.; Tu, Y.; Wu, Y.; Xie, E.; Yu, C.; Sun, W.; Li, X.; Zheng, J.; et al. Ischemia preconditioning alleviates ischemia/reperfusion injury-induced coronary no-reflow and contraction of microvascular pericytes in rats. Microvasc. Res. 2022, 142, 104349.
- Canty, J.M., Jr.; Klocke, F.J. Reduced regional myocardial perfusion in the presence of pharmacologic vasodilator reserve. Circulation 1985, 71, 370–377.
- VanBenthuysen, K.M.; McMurtry, I.F.; Horwitz, L.D. Reperfusion after acute coronary occlusion in dogs impairs endothelium-dependent relaxation to acetylcholine and augments contractile reactivity in vitro. J. Clin. Investig. 1987, 79, 265–274.
- Quillen, J.E.; Sellke, F.W.; Brooks, L.A.; Harrison, D.G. Ischemia-reperfusion impairs endothelium-dependent relaxation of coronary microvessels but does not affect large arteries. Circulation 1990, 82, 586–594.
- Malliani, A.; Schwartz, P.J.; Zanchetti, A. A sympathetic reflex elicited by experimental coronary occlusion. Am. J. Physiol. 1969, 217, 703–709.
- Gregorini, L.; Marco, J.; Kozakova, M.; Palombo, C.; Anguissola, G.B.; Marco, I.; Bernies, M.; Cassagneau, B.; Distante, A.; Bossi, I.M.; et al. Alpha-adrenergic blockade improves recovery of myocardial perfusion and function after coronary stenting in patients with acute myocardial infarction. Circulation 1999, 99, 482–490.
- Shimizu, M.; Wang, Q.D.; Sjoquist, P.O.; Ryden, L. The angiotensin II AT1-receptor antagonist candesartan improves functional recovery and reduces the no-reflow area in reperfused ischemic rat hearts. J. Cardiovasc. Pharmacol. 1999, 34, 78–81.
- Ryckwaert, F.; Colson, P.; Guillon, G.; Foex, P. Cumulative effects of AT1 and AT2 receptor blockade on ischaemia-reperfusion recovery in rat hearts. Pharmacol. Res. 2005, 51, 497–502.
- Gao, X.; Zhang, H.; Belmadani, S.; Wu, J.; Xu, X.; Elford, H.; Potter, B.J.; Zhang, C. Role of TNF-alpha-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2242–H2249.
- Kleinbongard, P.; Bose, D.; Baars, T.; Mohlenkamp, S.; Konorza, T.; Schoner, S.; Elter-Schulz, M.; Eggebrecht, H.; Degen, H.; Haude, M.; et al. Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ. Res. 2011, 108, 344–352.
- Kleinbongard, P.; Baars, T.; Mohlenkamp, S.; Kahlert, P.; Erbel, R.; Heusch, G. Aspirate from human stented native coronary arteries vs. saphenous vein grafts: More endothelin but less particulate debris. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1222–H1229.
- Herring, N.; Tapoulal, N.; Kalla, M.; Ye, X.; Borysova, L.; Lee, R.; Dall’Armellina, E.; Stanley, C.; Ascione, R.; Lu, C.J.; et al. Neuropeptide-Y causes coronary microvascular constriction and is associated with reduced ejection fraction following ST-elevation myocardial infarction. Eur. Heart J. 2019, 40, 1920–1929.
- Xue, H.M.; He, G.W.; Huang, J.H.; Yang, Q. New strategy of endothelial protection in cardiac surgery: Use of enhancer of endothelial nitric oxide synthase. World J. Surg. 2010, 34, 1461–1469.
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551.
- Hein, T.W.; Zhang, C.; Wang, W.; Chang, C.I.; Thengchaisri, N.; Kuo, L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: Counteracting role of arginase. FASEB J. 2003, 17, 2328–2330.
- Schreckenberg, R.; Weber, P.; Cabrera-Fuentes, H.A.; Steinert, I.; Preissner, K.T.; Bencsik, P.; Sarkozy, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; et al. Mechanism and consequences of the shift in cardiac arginine metabolism following ischaemia and reperfusion in rats. Thromb. Haemost. 2015, 113, 482–493.
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 864946.
- Viehman, G.E.; Ma, X.L.; Lefer, D.J.; Lefer, A.M. Time course of endothelial dysfunction and myocardial injury during coronary arterial occlusion. Am. J. Physiol. 1991, 261, H874–H881.
- Hollander, M.R.; de Waard, G.A.; Konijnenberg, L.S.; Meijer-van Putten, R.M.; van den Brom, C.E.; Paauw, N.; de Vries, H.E.; van de Ven, P.M.; Aman, J.; Van Nieuw-Amerongen, G.P.; et al. Dissecting the Effects of Ischemia and Reperfusion on the Coronary Microcirculation in a Rat Model of Acute Myocardial Infarction. PLoS ONE 2016, 11, e0157233.
- Gavin, J.B.; Seelye, R.N.; Nevalainen, T.J.; Armiger, L.C. The effect of ischaemia on the function and fine structure of the microvasculature of myocardium. Pathology 1978, 10, 103–111.
- Lonborg, J.; Kelbaek, H.; Helqvist, S.; Holmvang, L.; Jorgensen, E.; Saunamaki, K.; Klovgaard, L.; Kaltoft, A.; Botker, H.E.; Lassen, J.F.; et al. The impact of distal embolization and distal protection on long-term outcome in patients with ST elevation myocardial infarction randomized to primary percutaneous coronary intervention--results from a randomized study. Eur. Heart J. Acute Cardiovasc. Care 2015, 4, 180–188.
- Napodano, M.; Peluso, D.; Marra, M.P.; Frigo, A.C.; Tarantini, G.; Buja, P.; Gasparetto, V.; Fraccaro, C.; Isabella, G.; Razzolini, R.; et al. Time-dependent detrimental effects of distal embolization on myocardium and microvasculature during primary percutaneous coronary intervention. JACC Cardiovasc. Interv. 2012, 5, 1170–1177.
- Henriques, J.P.; Zijlstra, F.; Ottervanger, J.P.; de Boer, M.J.; van ‘t Hof, A.W.; Hoorntje, J.C.; Suryapranata, H. Incidence and clinical significance of distal embolization during primary angioplasty for acute myocardial infarction. Eur. Heart J. 2002, 23, 1112–1117.
- Yameogo, N.V.; Guenancia, C.; Porot, G.; Stamboul, K.; Richard, C.; Gudjoncik, A.; Hamblin, J.; Buffet, P.; Lorgis, L.; Cottin, Y. Predictors of angiographically visible distal embolization in STEMI. Herz 2020, 45, 288–292.
- Stone, G.W.; Webb, J.; Cox, D.A.; Brodie, B.R.; Qureshi, M.; Kalynych, A.; Turco, M.; Schultheiss, H.P.; Dulas, D.; Rutherford, B.D.; et al. Distal microcirculatory protection during percutaneous coronary intervention in acute ST-segment elevation myocardial infarction: A randomized controlled trial. JAMA 2005, 293, 1063–1072.
- Kleinbongard, P.; Heusch, G. A fresh look at coronary microembolization. Nat. Rev. Cardiol. 2022, 19, 265–280.
- Yunoki, K.; Naruko, T.; Inoue, T.; Sugioka, K.; Inaba, M.; Iwasa, Y.; Komatsu, R.; Itoh, A.; Haze, K.; Yoshiyama, M.; et al. Relationship of thrombus characteristics to the incidence of angiographically visible distal embolization in patients with ST-segment elevation myocardial infarction treated with thrombus aspiration. JACC Cardiovasc. Interv. 2013, 6, 377–385.
- Abela, G.S.; Kalavakunta, J.K.; Janoudi, A.; Leffler, D.; Dhar, G.; Salehi, N.; Cohn, J.; Shah, I.; Karve, M.; Kotaru, V.P.K.; et al. Frequency of Cholesterol Crystals in Culprit Coronary Artery Aspirate During Acute Myocardial Infarction and Their Relation to Inflammation and Myocardial Injury. Am. J. Cardiol. 2017, 120, 1699–1707.
- Katayama, Y.; Taruya, A.; Kashiwagi, M.; Ozaki, Y.; Shiono, Y.; Tanimoto, T.; Yoshikawa, T.; Kondo, T.; Tanaka, A. No-reflow phenomenon and in vivo cholesterol crystals combined with lipid core in acute myocardial infarction. Int. J. Cardiol. Heart Vasc. 2022, 38, 100953.
- Kawaguchi, R.; Oshima, S.; Jingu, M.; Tsurugaya, H.; Toyama, T.; Hoshizaki, H.; Taniguchi, K. Usefulness of virtual histology intravascular ultrasound to predict distal embolization for ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2007, 50, 1641–1646.
- Napodano, M.; Ramondo, A.; Tarantini, G.; Peluso, D.; Compagno, S.; Fraccaro, C.; Frigo, A.C.; Razzolini, R.; Iliceto, S. Predictors and time-related impact of distal embolization during primary angioplasty. Eur. Heart J. 2009, 30, 305–313.
- Fokkema, M.L.; Vlaar, P.J.; Svilaas, T.; Vogelzang, M.; Amo, D.; Diercks, G.F.; Suurmeijer, A.J.; Zijlstra, F. Incidence and clinical consequences of distal embolization on the coronary angiogram after percutaneous coronary intervention for ST-elevation myocardial infarction. Eur. Heart J. 2009, 30, 908–915.
- Falk, E.; Thuesen, L. Pathology of coronary microembolisation and no reflow. Heart 2003, 89, 983–985.
- Skyschally, A.; Walter, B.; Heusch, G. Coronary microembolization during early reperfusion: Infarct extension, but protection by ischaemic postconditioning. Eur. Heart J. 2013, 34, 3314–3321.
- Carlsson, M.; Martin, A.J.; Ursell, P.C.; Saloner, D.; Saeed, M. Magnetic resonance imaging quantification of left ventricular dysfunction following coronary microembolization. Magn. Reson. Med. 2009, 61, 595–602.
- Tucker, B.; Vaidya, K.; Cochran, B.J.; Patel, S. Inflammation during Percutaneous Coronary Intervention-Prognostic Value, Mechanisms and Therapeutic Targets. Cells 2021, 10, 1391.
- Quinones, A.; Saric, M. The cholesterol emboli syndrome in atherosclerosis. Curr. Atheroscler. Rep. 2013, 15, 315.
- Thomas, R.M.; Lim, S.Y.; Qiang, B.; Osherov, A.B.; Ghugre, N.R.; Noyan, H.; Qi, X.; Wolff, R.; Ladouceur-Wodzak, M.; Berk, T.A.; et al. Distal coronary embolization following acute myocardial infarction increases early infarct size and late left ventricular wall thinning in a porcine model. J. Cardiovasc. Magn. Reson. 2015, 17, 106.
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78.
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100.
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722.
- Kloner, R.A.; Alker, K.J. The Effect of Streptokinase on Intramyocardial Hemorrhage, Infarct Size, and the No-Reflow Phenomenon during Coronary Reperfusion. Circulation 1984, 70, 513–521.
- Ndrepepa, G.; Tiroch, K.; Keta, D.; Fusaro, M.; Seyfarth, M.; Pache, J.; Mehilli, J.; Schoemig, A.; Kastrati, A. Predictive Factors and Impact of No Reflow after Primary Percutaneous Coronary Intervention in Patients with Acute Myocardial Infarction. Circ. Cardiovasc. Interv. 2010, 3, 27–33.
- Kloner, R.A.; Rude, R.E.; Carlson, N.; Maroko, P.R.; DeBoer, L.W.; Braunwald, E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: Which comes first? Circulation 1980, 62, 945–952.
- Kloner, R.A.; King, K.S.; Harrington, M.G. No-reflow phenomenon in the heart and brain. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H550–H562.
- Ambrosio, G.; Weisman, H.F.; Mannisi, J.A.; Becker, L.C. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation 1989, 80, 1846–1861.
- Rochitte, C.E.; Lima, J.A.; Bluemke, D.A.; Reeder, S.B.; McVeigh, E.R.; Furuta, T.; Becker, L.C.; Melin, J.A. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 1998, 98, 1006–1014.
- Reffelmann, T.; Kloner, R.A. Microvascular reperfusion injury: Rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1099–H1107.
- Galiuto, L.; Lombardo, A.; Maseri, A.; Santoro, L.; Porto, I.; Cianflone, D.; Rebuzzi, A.G.; Crea, F. Temporal evolution and functional outcome of no reflow: Sustained and spontaneously reversible patterns following successful coronary recanalisation. Heart 2003, 89, 731–737.
- Turschner, O.; D’Hooge, J.; Dommke, C.; Claus, P.; Verbeken, E.; De Scheerder, I.; Bijnens, B.; Sutherland, G.R. The sequential changes in myocardial thickness and thickening which occur during acute transmural infarction, infarct reperfusion and the resultant expression of reperfusion injury. Eur. Heart J. 2004, 25, 794–803.
- Fernandez-Jimenez, R.; Sanchez-Gonzalez, J.; Aguero, J.; Garcia-Prieto, J.; Lopez-Martin, G.J.; Garcia-Ruiz, J.M.; Molina-Iracheta, A.; Rossello, X.; Fernandez-Friera, L.; Pizarro, G.; et al. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: Imaging and histological tissue characterization. J. Am. Coll. Cardiol. 2015, 65, 315–323.
- Alkhalil, M.; Borlotti, A.; De Maria, G.L.; Gaughran, L.; Langrish, J.; Lucking, A.; Ferreira, V.; Kharbanda, R.K.; Banning, A.P.; Channon, K.M.; et al. Dynamic changes in injured myocardium, very early after acute myocardial infarction, quantified using T1 mapping cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2018, 20, 82.
- Fernandez-Jimenez, R.; Barreiro-Perez, M.; Martin-Garcia, A.; Sanchez-Gonzalez, J.; Aguero, J.; Galan-Arriola, C.; Garcia-Prieto, J.; Diaz-Pelaez, E.; Vara, P.; Martinez, I.; et al. Dynamic Edematous Response of the Human Heart to Myocardial Infarction: Implications for Assessing Myocardial Area at Risk and Salvage. Circulation 2017, 136, 1288–1300.
- Veinot, J.P.; Gattinger, D.A.; Fliss, H. Early apoptosis in human myocardial infarcts. Hum. Pathol. 1997, 28, 485–492.
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417.
- Kidambi, A.; Mather, A.N.; Motwani, M.; Swoboda, P.; Uddin, A.; Greenwood, J.P.; Plein, S. The effect of microvascular obstruction and intramyocardial hemorrhage on contractile recovery in reperfused myocardial infarction: Insights from cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2013, 15, 58.
- Kandler, D.; Lucke, C.; Grothoff, M.; Andres, C.; Lehmkuhl, L.; Nitzsche, S.; Riese, F.; Mende, M.; de Waha, S.; Desch, S.; et al. The relation between hypointense core, microvascular obstruction and intramyocardial haemorrhage in acute reperfused myocardial infarction assessed by cardiac magnetic resonance imaging. Eur. Radiol. 2014, 24, 3277–3288.
- Higginson, L.A.; White, F.; Heggtveit, H.A.; Sanders, T.M.; Bloor, C.M.; Covell, J.W. Determinants of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 1982, 65, 62–69.
- Mathey, D.G.; Schofer, J.; Kuck, K.H.; Beil, U.; Kloppel, G. Transmural, haemorrhagic myocardial infarction after intracoronary streptokinase. Clinical, angiographic, and necropsy findings. Br. Heart J. 1982, 48, 546–551.
- McNamara, J.J.; Lacro, R.V.; Yee, M.; Smith, G.T. Hemorrhagic infarction and coronary reperfusion. J. Thorac. Cardiovasc. Surg. 1981, 81, 498–501.
- Lotan, C.S.; Bouchard, A.; Cranney, G.B.; Bishop, S.P.; Pohost, G.M. Assessment of postreperfusion myocardial hemorrhage using proton NMR imaging at 1.5 T. Circulation 1992, 86, 1018–1025.
- Adamson, R.H.; Zeng, M.; Adamson, G.N.; Lenz, J.F.; Curry, F.E. PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H406–H417.
- Inauen, W.; Granger, D.N.; Meininger, C.J.; Schelling, M.E.; Granger, H.J.; Kvietys, P.R. Anoxia-reoxygenation-induced, neutrophil-mediated endothelial cell injury: Role of elastase. Am. J. Physiol. 1990, 259, H925–H931.
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197.
- Tarikuz Zaman, A.K.; Spees, J.L.; Sobel, B.E. Attenuation of cardiac vascular rhexis: A promising therapeutic target. Coron. Artery Dis. 2013, 24, 245–252.
- Zeman, A.K.M.T.; French, C.J.; Spees, J.L.; Binbrek, A.S.; Sobel, B.E. Vascular rhexis in mice subjected to non-sustained myocardial ischemia and its therapeutic implications. Exp. Biol. Med. 2011, 236, 598–603.
- Robbers, L.F.; Eerenberg, E.S.; Teunissen, P.F.; Jansen, M.F.; Hollander, M.R.; Horrevoets, A.J.; Knaapen, P.; Nijveldt, R.; Heymans, M.W.; Levi, M.M.; et al. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur. Heart J. 2013, 34, 2346–2353.
- Carrick, D.; Haig, C.; Ahmed, N.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M.M.; Davie, A.; et al. Myocardial Hemorrhage after Acute Reperfused ST-Segment-Elevation Myocardial Infarction: Relation to Microvascular Obstruction and Prognostic Significance. Circ. Cardiovasc. Imaging 2016, 9, e004148.
- Bulluck, H.; Rosmini, S.; Abdel-Gadir, A.; White, S.K.; Bhuva, A.N.; Treibel, T.A.; Fontana, M.; Ramlall, M.; Hamarneh, A.; Sirker, A.; et al. Residual Myocardial Iron Following Intramyocardial Hemorrhage During the Convalescent Phase of Reperfused ST-Segment-Elevation Myocardial Infarction and Adverse Left Ventricular Remodeling. Circ. Cardiovasc. Imaging 2016, 9, e004940.
- Kali, A.; Kumar, A.; Cokic, I.; Tang, R.L.; Tsaftaris, S.A.; Friedrich, M.G.; Dharmakumar, R. Chronic manifestation of postreperfusion intramyocardial hemorrhage as regional iron deposition: A cardiovascular magnetic resonance study with ex vivo validation. Circ. Cardiovasc. Imaging 2013, 6, 218–228.
- Waller, B.F.; Rothbaum, D.A.; Pinkerton, C.A.; Cowley, M.J.; Linnemeier, T.J.; Orr, C.; Irons, M.; Helmuth, R.A.; Wills, E.R.; Aust, C. Status of the myocardium and infarct-related coronary artery in 19 necropsy patients with acute recanalization using pharmacologic (streptokinase, r-tissue plasminogen activator), mechanical (percutaneous transluminal coronary angioplasty) or combined types of reperfusion therapy. J. Am. Coll. Cardiol. 1987, 9, 785–801.
- Amier, R.P.; Tijssen, R.Y.G.; Teunissen, P.F.A.; Fernandez-Jimenez, R.; Pizarro, G.; Garcia-Lunar, I.; Bastante, T.; van de Ven, P.M.; Beek, A.M.; Smulders, M.W.; et al. Predictors of Intramyocardial Hemorrhage after Reperfused ST-Segment Elevation Myocardial Infarction. J. Am. Heart Assoc. 2017, 6, e005651.
- Liu, T.; Howarth, A.G.; Chen, Y.; Nair, A.R.; Yang, H.J.; Ren, D.; Tang, R.; Sykes, J.; Kovacs, M.S.; Dey, D.; et al. Intramyocardial Hemorrhage and the “Wave Front” of Reperfusion Injury Compromising Myocardial Salvage. J. Am. Coll. Cardiol. 2022, 79, 35–48.
- Wu, K.C.; Zerhouni, E.A.; Judd, R.M.; Lugo-Olivieri, C.H.; Barouch, L.A.; Schulman, S.P.; Blumenthal, R.S.; Lima, J.A. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998, 97, 765–772.
- Kloner, R.A.; Giacomelli, F.; Alker, K.J.; Hale, S.L.; Matthews, R.; Bellows, S. Influx of neutrophils into the walls of large epicardial coronary arteries in response to ischemia/reperfusion. Circulation 1991, 84, 1758–1772.
- Hansen, P.R. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995, 91, 1872–1885.
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214.
- Montone, R.A.; La Vecchia, G. Interplay between inflammation and microvascular obstruction in ST-segment elevation myocardial infarction: The importance of velocity. Int. J. Cardiol. 2021, 339, 7–9.
- Mayr, A.; Klug, G.; Schocke, M.; Trieb, T.; Mair, J.; Pedarnig, K.; Pachinger, O.; Jaschke, W.; Metzler, B. Late microvascular obstruction after acute myocardial infarction: Relation with cardiac and inflammatory markers. Int. J. Cardiol. 2012, 157, 391–396.
- Reindl, M.; Reinstadler, S.J.; Feistritzer, H.J.; Klug, G.; Tiller, C.; Mair, J.; Mayr, A.; Jaschke, W.; Metzler, B. Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2017, 6, 640–649.
- Guo, F.; Dong, M.; Ren, F.; Zhang, C.; Li, J.; Tao, Z.; Yang, J.; Li, G. Association between local interleukin-6 levels and slow flow/microvascular dysfunction. J. Thromb. Thrombolysis 2014, 37, 475–482.
- Shetelig, C.; Limalanathan, S.; Hoffmann, P.; Seljeflot, I.; Gran, J.M.; Eritsland, J.; Andersen, G.O. Association of IL-8 with Infarct Size and Clinical Outcomes in Patients with STEMI. J. Am. Coll. Cardiol. 2018, 72, 187–198.
- Funayama, H.; Ishikawa, S.E.; Sugawara, Y.; Kubo, N.; Momomura, S.; Kawakami, M. Myeloperoxidase may contribute to the no-reflow phenomenon in patients with acute myocardial infarction. Int. J. Cardiol. 2010, 139, 187–192.
- Stamboul, K.; Zeller, M.; Rochette, L.; Cottin, Y.; Cochet, A.; Leclercq, T.; Porot, G.; Guenancia, C.; Fichot, M.; Maillot, N.; et al. Relation between high levels of myeloperoxidase in the culprit artery and microvascular obstruction, infarct size and reverse remodeling in ST-elevation myocardial infarction. PLoS ONE 2017, 12, e0179929.

