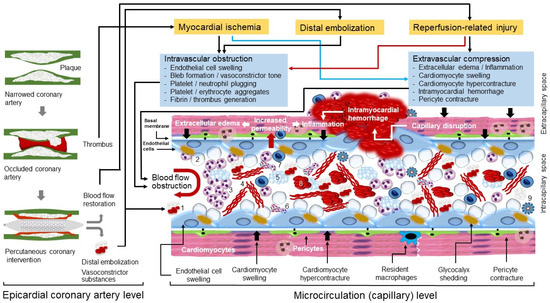
2. Pathophysiology of CNR
Microvascular obstruction (MVO) is the underlying pathophysiological mechanism of CNR. MVO and CNR after reperfusion of an occluded coronary artery are explained by a joint action of at least four factors: myocardial ischemia, spontaneous or iatrogenic distal embolization, reperfusion-related injury and individual susceptibility (predisposing conditions that increase the odds of developing MVO and CNR). Pathophysiological mechanisms of MVO and CNR are shown in
Figure 1.
Figure 1. Pathophysiological mechanisms of microvascular obstruction and coronary no-reflow. Pathophysiological events that develop at epicardial coronary artery and microcirculation (capillary) levels are shown. Black arrows denote forces generated outside the capillaries that compress microcirculation, the red arrow points out to increased permeability. Numbers show the following components of microvascular obstruction: 1, atherothrombotic debris; 2, blebs; 3, neutrophil aggregates; 4, fibrin; 5, platelet aggregates; 6, neutrophil extracellular traps (NETs); 7, monocytes; 8; erythrocyte aggregates; 9, inflammasome.
2.1. A Short Description of Myocardial Microcirculation
Coronary microcirculation refers to blood circulation in vessels <200 µm in diameter that are not visualized on coronary angiography. It consists of arterioles, capillaries and venules. All 3 structures participate in the MVO and CNR following ischemia and reperfusion, albeit with different roles. Coronary arterioles have a relatively thick smooth muscle wall, act as resistance vessels and are responsible for keeping a constant precapillary pressure of ~45 mmHg in the setting of autoregulation. In the setting of ischemia/reperfusion injury arterioles contribute to MVO and CNR through impaired vasomotor tone (impaired endothelium dependent vasodilation) and propensity to in situ thrombus formation. Morphometric analyses have shown that there are approximately 2200 capillaries per square millimeter in adult human hearts
[23]. It has been estimated that there are approximately 8 million capillaries in the human heart. Coronary capillaries contain ~1/3 of myocardial blood (~45 mL) which moves with an average speed of 1 mm/sec (at resting state) under a hydrostatic pressure of ~30 mmHg
[24][25][24,25]. In the setting of ischemia/reperfusion injury, coronary capillaries undergo constriction, obstruction, and compression, which lead to a marked reduction in the number of open vessels and diminished delivery of oxygen and nutrients to the surrounding tissue. Coronary capillaries are the main site of clogged microcirculation and MVO occurring as a response to ischemia and/or reperfusion. Coronary venules have a weak smooth muscle and, consequently, manifest weak muscular vascular responses (venular hydrostatic pressure is ~15 mmHg). However, coronary venules participate in the ischemia and/or reperfusion-related MVO by serving as a preferential site of leucocyte and platelet adhesion via expression of adhesion molecules and subsequent inflammation
[25][26][25,26].
2.2. Myocardial Ischemia
Total cessation or drastic reduction (>80%) of coronary blood flow results in severe myocardial ischemia in subtended myocardium. Previous studies that have investigated cardioprotective measures in ischemia/reperfusion models were potentially conceptually flawed in that they focused on the cardiomyocytes paying little attention to microcirculation. This could be one reason of the failure of cardioprotective measures to translate into clinical benefit
[27]. Microcirculation—the key component of MVO and CNR—is vulnerable to ischemia and reperfusion injury. In the following material,
reswe
archers focused on the endothelial cells and other components of microcirculation, whereas the impact of ischemia on cardiomyocytes was not addressed.
Endothelial cells are more resistant to ischemia than surrounding cardiomyocytes and may survive hypoxia for minutes to days following the installation of ischemia
[10][28][10,28]. Endothelial cells are abundant in myocardium representing 3% to 5% of the myocardial volume or approximately 45% of total cells or 60% of nonmyocyte cells in the murine myocardium
[29]. Experimental studies using human umbilical vein endothelial cells showed that 75% of cells survived 24 h of hypoxia
[30] and >50% of cells survived 48 h of hypoxia
[28]. Concentration of high-energy phosphates (adenosine triphosphate [ATP] and creatine phosphate) is low in myocardium and can support contraction for only a few effective systoles. The oxygen present in capillaries (as oxyhemoglobin) and cardiomyocytes (as oxymyoglobin) is exhausted after 8–10 s, which brings to almost total cessation of oxidative phosphorylation and effective myocardial contraction. At 15 to 20 s of ischemia (artery occlusion), anaerobic glycolysis supervenes as the only source of generation of ATP. At 60 s of ischemia anaerobic glycolysis slows markedly and at 40 to 60 min of total ischemia, anaerobic glycolysis essentially stops
[31]. The lack of coronary blood flow and cessation of aerobic metabolism lead to accumulation of various catabolites in the cells and interstitial space of ischemic tissue including lactate, protons (tissue acidosis), ammonium, degraded nucleotide phosphates (adenosine diphosphate, adenosine), products of glycogen breakdown (glucose-1-phosphate), glucose-6-phosphate and many other products of intermediary metabolism. Higher concentrations of these catabolites increase the osmotic load within the cells and interstitial space, which generates an osmotic gradient forcing the water to move inside the cells or from intracapillary to interstitial space leading to cellular swelling and interstitial edema
[32]. Thus, endothelial cell swelling and interstitial edema are important contributors to MVO and CNR during myocardial ischemia.
Lack of high-energy phosphates leads to severe perturbations in ionic hemostasis in endothelial cells. Apart from further contributing to endothelial cell swelling and intracapillary space obstruction as a response to ischemia, altered ionic hemostasis has other negative actions promoting endothelial cell dysfunction. Ischemic endothelial cells show increased concentration of calcium in cytoplasm, which activates endothelial cell contractile elements
[33]. Actin filaments constitute 5–15% of the total protein in endothelial cells
[34] and actin cytoskeleton is critical for maintenance of endothelial barrier function
[35]. Calcium-induced contraction of endothelial cell filaments reduces mechanical support for endothelial cell membrane promoting cytoplasmic budding or blebbing into the intracapillary space
[36]. Bleb formation appears to be further favored by loss of antegrade pulsatile flow and increased shear stress
[37]. Blebs and their role in the intracapillary obstruction have been described since inaugural structural studies of no-reflow
[2][3][2,3]. Calcium-induced filament contraction appear to change the cellular shape, which destabilizes cellular junctions and increase intercellular permeability. There are other factors that destabilize endothelial intercellular junctions during myocardial ischemia. Ischemia-induced expression of vascular endothelial growth factor (VEGF)—a major regulator of vascular permeability
[38]—and dissociation of VEGF receptor 2-vascular endothelial (VE)-cadherin (a cell adhesion protein of adherens junctions) complex lead to increased inter-endothelial cell permeability
[39]. VEGF activates Scr (a member of Src kinase family) via phosphorylation, which leads to phosphorylation of tyrosine residues of VE-cadherin of the interendothelial cell junctions. This action promotes VE-cadherin internalization and reduces the amount of VE-cadherin in the interendothelial cell junctions
[40][41][40,41]. The removal of VE-cadherin from the intercellular junctions further destabilizes intercellular connection and increase intercellular permeability. In experimental conditions VE-cadherin phosphorylation is also facilitated by increased shear stress
[40]. VEGF also activates endothelial nitric oxide synthase (eNOS) in the caveolae of endothelial cells
[42], further contributing to increased vascular permeability. Activated endothelial and circulating cells (platelets and neutrophils) show increased expression of adhesion molecules
[43], which may be further potentiated by subsequent reperfusion by thrombolysis or angioplasty
[44]. Exposed adhesion molecules mediate platelet and leukocyte endothelial interactions, which facilitate trapping of these cells in the ischemic microvascular space (discussed later under this subheading).
Glycocalyx is perhaps the earliest microcirculation component that is damaged in the course of ischemia/reperfusion. Glycocalyx is an important component of endothelial barrier. Glycocalyx represents a 0.5 µm thick carbohydrate-rich matrix that covers the endothelium surface throughout the capillary system
[45]. The thickness of glycocalyx exceeds the length of extracellular domains of most endothelial adhesion molecules
[46], which in normal conditions prevents the adhesion of circulating cells to endothelial cells. The highly hydrophilic nature of glycocalyx enables creation of a relatively fixed (albeit exchangeable) water layer on the surface of endothelial cells, which together with electrostatic interactions with circulating erythrocytes, reduces the capillary hematocrit compared with that found in the systemic circulation and facilitates the passage of blood through the capillaries
[47]. Glycocalyx is degraded upon exposure to ischemia
[48][49][48,49], reactive oxygen species (ROS)
[49][50][49,50], oxidized lipoproteins
[51], acute hyperglycemia
[52], tumor necrosis factor alpha (TNFα)
[53], matrix metalloproteinase (MMP) 2 and 9
[54], inflammatory states and vigorous volume loading
[55]. Nitric oxide (NO) appears to be protective against glycocalyx shedding
[56]. Glycocalyx degradation (shedding) contributes to MVO and CNR by damaging the capillary barrier and increasing capillary permeability, which contributes to endothelial cell and interstitial edema
[57] and by enabling leukocyte
[58] and platelet
[59] adhesion to endothelial cells facilitating the entrapment of these cells in the intracapillary space.
Platelets and neutrophils are recruited in the capillaries following myocardial ischemia and contribute to ischemic injury, MVO and CNR
[60]. Following activation by ischemia, platelets expose their adhesion molecules and aggregate to endothelial cells (facilitated by glycocalyx shedding), neutrophils, erythrocytes and to each other contributing to microcirculation obstruction (
Figure 1). These cellular aggregates have been demonstrated in capillaries from the very first ultramicroscopic characterization of no-reflow
[2][3][2,3]. A significant increase in the neutrophil–platelet aggregates and monocyte–platelet aggregates was shown in the coronary sinus blood samples of microsphere-induced CNR in Yorkshire pigs
[61]. Apart from mechanical blockage by aggregates and microthrombi, activated platelets release various biological active substances including nucleotides, proteases, platelet-activating factor (PAF), ROS, adhesive proteins (fibronectin, von Willebrand factor, thrombospondin, P-selectin, glycoprotein IIb/IIIa, fibrinogen), vasoconstrictors such as thromboxane A2 and serotonin, growth factors, coagulation and complement system factors, various cytokines and chemokines, pro-angiogenic factors and microvesicles and exosomes
[60][62][63][64][65][66][60,62,63,64,65,66]. These substances contribute to proteolytic destruction of endothelial cells and intercellular junctions (and increased permeability), intracapillary blood coagulation, chemiotaxis and recruitment of leukocytes in microcirculation and promote inflammation, apoptosis and angiogenesis. However, it appears that the degree and nature of platelet contribution to ischemia and reperfusion-related injury depend on the state of platelet activation. Experimental studies in rats and guinea pigs showed that ischemia/reperfusion damage was ameliorated and endothelial integrity was improved by platelet or platelet-derived perfusion
[67][68][67,68]. Platelet glycoprotein IIb/IIIa receptor blockage has reduced microvascular thrombosis in murine models of acute stroke
[69] and platelet depletion counteracts deleterious effects of acute hypercholesterolemia on infarct size and CNR in an ischemia/reperfusion model in rabbits
[70]. Glycoprotein IIb/IIIa receptor blockade with abciximab improved the recovery of microvascular perfusion and enhanced the recovery of contractile function in the area at risk in patients with AMI after coronary stenting
[71]. Neutrophils are recruited early in the ischemic myocardium
[60]. Neutrophils transmigrate through endothelial cells by interaction with endothelial cell junction proteins due to the highly chemotactic milieu in the ischemic microcirculation
[72]. Activated neutrophils aggregate with other cells and form neutrophil extracellular traps (NETs) clogging the microcirculation and impeding blood flow
[73][74][73,74]. Neutrophils are a major source of ROS
[75], myeloperoxidase
[76] and proteolytic enzymes (such as, elastase and metalloproteinase-9)
[77][78][77,78], which in turn, promote degradation of all components of capillary barrier (glycocalyx, endothelial cells and basal membrane), leading to vascular leakage, increased vascular permeability and excess edema. Following initial infiltration and activation, neutrophils and other inflammatory cells (monocyte/macrophages and lymphocytes) participate in the powerful local and systemic inflammatory response that develops in patients with AMI.
Growing evidence suggests that pericytes play an important role in the genesis of CNR. With a density of approximately 3.6 × 10
7 pericytes/cm
3, pericyte is the second most frequent nonmyogenic cell found in the heart in vitro
[79]. Pericytes contract in circumferential and longitudinal directions influencing the diameter and stiffness of capillaries. The close proximity of pericytes to sympathetic axons suggests that their tone may be under noradrenergic regulation
[80]. Pericytes have an established role in autoregulation of cerebral blood flow and contribute to vasoconstriction of cerebral capillaries and entrapment of erythrocytes and leukocytes in no-reflow zones following cerebral ischemia
[81]. In mouse models of cerebral ischemia, pericytes caused capillary constriction and obstructed erythrocyte passage despite reopening the middle cerebral artery
[82]. Cardiac pericytes constrict coronary capillaries and reduce microvascular blood flow after ischemia, despite reopening of the culprit artery
[80]. In rat models of ischemia and reperfusion, areas of capillary blockage colocalized with pericytes, which showed a 37% diameter reduction. Notably, intravenous adenosine—a pericyte relaxant drug—increased the capillary diameter by 21% (at pericyte somata), decreased the capillary block by 25% and increased the perfusion volume by 57%
[80]. Recent evidence also strongly suggests that pericytes contract as a response to myocardial ischemia and they play an important role in CNR
[83][84][83,84]. Ischemic preconditioning inhibited the contraction of microvascular pericytes induced by cardiac ischemia/reperfusion injury suggesting that protective role of ischemic preconditioning may be at least partially mediated by its impact on pericyte function
[85]. It has been proposed that cardiac pericytes may represent a novel therapeutic target aiming at protection of coronary microcirculation and alleviation of MVO and CNR after AMI
[65].
Myocardial ischemia sets into operation a large number of systemic and local vasoconstrictor stimuli that impair the coronary vasodilator reserve and increase the vasoconstrictor tone of microcirculation, which may be alleviated by vasodilator drug therapy
[86]. Experimental studies have shown that arterioles undergoing ischemia/reperfusion fail to dilate under the effect of endothelium dependent vasoactive substances acetylcholine and bradykinin, suggesting that endothelium-dependent relaxation of coronary microvessels was markedly impaired during ischemia/reperfusion cycle
[87][88][87,88]. Although the underlying mechanisms of persistently increased vasoconstrictor tone in microcirculation undergoing ischemia/reperfusion are unknown, excess alpha-adrenergic tone
[89][90][89,90], angiotensin II
[91][92][91,92], excessive production of ROS and cytokines-like tumor necrosis factor alpha (TNFα)
[93][94][93,94], vasoconstrictor substances released from the culprit lesions including serotonin and thromboxane A2
[94], endothelin
[95] or neuropeptide Y
[96], most likely in combination, do play a role. Reduced availability of NO caused by inhibition
[97] or uncoupling of endothelial nitric oxide synthase
[98], upregulation of arginase
[99][100][99,100] and increased production of ROS
[101] affect vascular tone (among other deleterious effects) during ischemia/reperfusion. Although increased vasoconstrictor tone was considered deleterious in that it contributes to obstructed microcirculation and MVO,
rwe
searchers hypothesize that increased vasoconstrictor tone in the ischemic area may have a protective role as well. By obstructing the microcirculation, the increased vasoconstrictor tone confines ischemic products, necrotic debris and a large number of harmful catabolites and active substances to the ischemic area, preventing them from spreading to surrounding viable myocardium or from entering the circulation. This may be at least one reason why vasodilator therapy fails to improve clinical outcome, although it apparently may improve reperfusion. In addition, vasodilator therapy may preferentially dilate vessels surrounding ischemic region and shift the blood towards viable surrounding myocardium (microcirculation steal syndrome) worsening the reperfusion of ischemic region, apparently associated with improved reperfusion, at least by angiographic (TIMI flow grade) markers. However, these hypotheses need testing.
In aggregate, myocardial ischemia leads to various alterations affecting all components of microcirculation leading to various degrees of MVO and CNR. Endothelial cells, however, are relatively resistant to ischemia and may remain ultrastructurally intact up to 6 h of ischemia in anesthetized cats
[102]. In rat models of AMI, no clear damage to capillary endothelium occurred after 30 min of ischemia without reperfusion and no reduction of inter-endothelial cell junctions was observed after 90 min occlusion of left anterior descending artery
[103]. Another study in mongrel dogs subjected to ligation of the circumflex branch showed mild swelling of endothelial cells but no totally occluded capillaries following prolonged periods of ischemia. The study suggested that ischemia-induced loss of vascular competence was unlikely to be due to intravascular thrombosis, endothelial cell swelling, or external compression by interstitial edema
[104]. Thus, shorter periods of ischemia (≤1 h) are characterized by mild edema with almost no (or little) signs of cellular necrosis, inflammation or capillary injury. Longer periods of ischemia (≥2 h) are characterized by a uniform infarcted area (cellular death), marked infiltration by inflammatory cells (primarily neutrophils), and severely damaged capillaries and evident hemorrhage
[54].
2.3. Distal Embolization
Distal embolization of atherothrombotic fragments from the atherosclerotic plaque occurs spontaneously or during the primary PCI procedure as a result of guide-wire passage, lesion preparation and stent implantation. Angiographically visible distal embolization is documented in 11% to 17% of primary PCI procedures in patients with STEMI
[105][106][107][108][105,106,107,108]. However, the true incidence of lesser degrees of distal embolization appears to be much higher, with one study showing visible debris in 73% of patients who received a distal embolization protection system
[109]. Histologically, the embolized material consists of a mixture of atheromatous debris, platelet aggregates, erythrocytes, fibrin, cholesterol crystals and inflammatory cells
[110][111][112][113][110,111,112,113]. Distal embolization is more frequent in atherosclerotic plaques with large volumes (particularly plaques with large necrotic core)
[114] and those with more thrombus at the lesion site
[115][116][115,116]. Moreover, erythrocyte-rich thrombi, elevated glucose level on admission, larger culprit vessel, pre-balloon dilation and right coronary artery as culprit lesion have been identified as independently associated with a higher risk of distal embolization during the primary PCI procedures
[108][111][108,111]. Although most studies have assessed distal embolization in patients with STEMI characterized by a large thrombus burden, it has been suggested that distal microembolization may occur during plaque erosion at the culprit lesion in patients presenting with non-STEMI
[110]. Since microthrombi preferentially end in well reperfused and viable myocardium (directed by blood stream), distal embolization kills potentially salvageable myocardium
[117]. One experimental study in dogs suggested that embolizing particles tend to flow away from the central infarcted area (forced by developing CNR) and accumulate in the infarct border contributing to infarct extension
[118]. The study suggested that embolizing particles are more important for infarct expansion than for CNR at least in the early phase of reperfusion. Distal embolization contributes to CNR, increases biomarkers of myocardial necrosis, causes patchy microinfarcts that disproportionally impair the left ventricular function beyond the actual amount of damaged myocardium, increases infarct size and is associated with a poor clinical outcome
[41][107][118][119][41,107,118,119]. Although CNR may develop in the absence of distal embolization, distal embolization may promote (or aggravate) MVO and CNR by a number of mechanisms (recently reviewed by Kleinbongard and Heusch
[110]) including mechanical obstruction, increased vasoconstrictor tone at arteriolar and microcirculation levels by soluble vasoconstrictor substances contained in embolized material
[94][95][96][94,95,96], generation of a prothrombotic milieu and favorization of platelet aggregation and in situ thrombosis
[110][120][110,120] and inducing a powerful local inflammatory response
[110][121][110,121]. Experimental studies of microthrombi-induced CNR in Yorkshire pigs showed that distal embolization was associated with elevated levels of metalloproteinase-2 and a reduction in the activity of survival kinase (Akt) within the infarct zone 3 days after AMI, with both events helping to explain deleterious effects of distal embolization on infarct size
[122]. Atheromatous debris fraction of the embolized material is extremely resistant to antithrombotic or thrombolytic agents used to treat patients with AMI and the only way to clear it is through inflammation or other processes set into operation to clear necrotic tissue after AMI.
2.4. Reperfusion-Related Injury
Endothelial cells and microcirculation are extremely sensitive to reperfusion-related injury—a condition first described by Jennings et al.
[123] in 1960 in canine hearts. There are at least five manifestations of reperfusion-related injury: reperfusion-induced arrhythmias, myocardial stunning, MVO, intra-myocardial hemorrhage and reperfusion-induced cell death (or lethal reperfusion injury)
[124][125][124,125]. Although, myocardial ischemia and reperfusion appear to be opposite events in terms of blood interruption and restoration, they are similar in terms of molecular and cellular events that develop following both events. It appears that reperfusion-related injury completes the cellular damage initiated by ischemia. However, the motif of reperfusion-related injury is unclear but it may represent an early scavenger mechanism to clear ischemia-induced irreversibly damaged cells.
Experimental studies in dogs by Kloner et al.
[10] showed that fluorescent dye thioflavin S managed to penetrate ischemic myocardium after 40 min of ischemia followed by reperfusion. However, after 90 min of ischemia followed by reperfusion, thiofalvin S failed to penetrate the ischemic myocardium and perfusion defects were observed in subendocardium. Reperfusion failure was observed within seconds of clamp release and it was well established within the first few minutes. Notably, perfusion defects were always found within the ischemic-necrotic zone but not in the surrounding viable myocardium not undergoing ischemia/reperfusion, strongly suggesting that CNR is due to microvascular damage within the zone of necrosis
[10]. Later studies showed that if proximal coronary arteries were occluded for a longer time (3 h), then perfusion defects were more widespread and reached mid-myocardium and occasionally the outer layers of myocardium
[126]. These studies showed that the extent of CNR depends on the duration of ischemia, a finding that has been confirmed in the clinical studies as well
[127]. The most consistent histologic finding was demonstration of areas of swollen endothelium and formation of intraluminal membrane-bound protrusions or blebs that obstructed the capillary lumen. Other (less frequently observed) markers of microcirculation damage included loss of pinocytotic vesicles, endothelial gaps, rupture of capillary walls with extravasation of red blood cells, deposits of fibrin tactoids in vicinity of endothelium gaps, platelet–leukocyte aggregates, and rouleaux structures of erythrocytes. The local edema involving endothelium and surrounding myocardium suggested an initial restoration of some blood flow, which was later interrupted by reperfusion-induced MVO and swollen cardiomyocytes. Occasionally a capillary compressed (and consequently obstructed) by two swollen cardiomyocytes was seen. Reperfusion-induced hypercontracture of myocardium was also involved in the compression of microcirculation
[128][129][128,129]. Studies by Ambrosio et al.
[130] in open-chest dogs subjected to 90 min occlusion of left circumflex coronary artery followed by reperfusion for 2 min or 3.5 h showed that the extent of the CNR area grows over the reperfusion time. Thus, the area of impaired reperfusion (absent thioflavin) was 9.5% of the initial area at risk in animals reperfused for 2 min and 25.9% of the area at risk in dogs reperfused for 3.5 h. Importantly, serial measurements using microspheres showed that areas with adequate reperfusion at 30 min of reperfusion had a marked fall of perfusion at 3.5 h of reperfusion. Another study in dogs undergoing 90 min (balloon) occlusion of the left anterior descending coronary artery followed by reflow showed that the extent of MVO (assessed by hypo-enhanced regions on contrast-enhanced cardiac magnetic resonance [CMR]) increased threefold over the 48 h after reperfusion (3.2%, 6.7% and 9.9% of the left ventricular mass at 2, 6, and 48 h, respectively)
[131]. Similar findings were reported by Reffelmann and Kloner
[132] in a rabbit model of reperfusion. The area of CNR increased progressively from 12.2% after 2 min of reperfusion to 30.8% after 2 h of reperfusion and to 34.9% of the initial area at risk after 8 h of reperfusion. The expansion of CNR zone was fastest within the first 2 h of reperfusion, finally encompassing ~80% of the infarct size. Regional myocardial blood flow was hyperemic at 2 min of reperfusion, decreased later and remained unchanged (plateau) between 2 and 8 h of reperfusion. Notably, no hemorrhage was visible after 2 min of reperfusion but it reached a value of 37.3% of area at risk after 8 h of reperfusion. One study that included patients with the first AMI showed that MCE-defined CNR present at 24 h after reperfusion was sustained at 1 month in approximately 50% of the patients
[133]. These studies strongly suggested that CNR is primarily a reperfusion injury-related phenomenon.
One of the earliest events that develop following blood flow restoration to ischemic myocardium is exacerbation of ischemia-initiated interstitial and cellular edema. An experimental study in dogs undergoing a 90 min balloon occlusion of left circumflex artery followed by 60 min reperfusion showed increased wall thickness in the reperfused myocardium due to tissue edema eventually leading to CNR because of mechanical compression
[134]. CMR imaging studies in pigs
[135] and humans
[136][137][136,137] have shown a bimodal pattern of myocardial edema following reperfusion. The early wave of edema appears to be due to exposure of a hyperosmotic interstitium (due to accumulation of catabolites produced during ischemia) to normo-osmotic blood at reperfusion. The early wave of edema occurs immediately after reperfusion and markedly diminished at 24 h as a result of catabolite washout from the interstitium
[135]. The second (late) wave of edema develops gradually following ischemia/reperfusion and is maximal around day 7 following reperfusion
[135]. The second wave of edema is explained by increased vascular permeability related to influx of inflammatory cells and healing process of the infarcted tissue
[135].
Ischemia-initiated endothelial cell edema is further accentuated by reperfusion. Upon restoration of blood flow, the extracellular pH is rapidly restored, which stimulates the Na
+/H
+ exchanger and Na
+/HCO3
- symporter leading to proton extrusion from the cells, rapid normalization of intracellular pH, massive Na
+ influx, and intracellular Ca
2+ overload
[36][124][36,124]. The increased Ca
2+ in endothelial cells leads to cellular retraction and intercellular gap formation and blebbing resulting in increased vascular permeability and obstruction of intracapillary space. In addition to cellular swelling, increased ATP availability upon restoration of blood flow, restored intracellular pH and abundant cytoplasmic Ca
2+ favor the hypercontracture of cardiomyocytes and contraction band formation—a histological marker of reperfusion
[138] further compressing the microvasculature. One of the most important deleterious effects of reperfusion is mitochondrial injury related to opening of mitochondrial permeability transition pore (MPTP)—a nonselective channel that enables movement of water, ions and low molecular weight solutes across the inner mitochondrial membrane. The MPTP channel remained closed during ischemia under the inhibitory effect of acidosis and increased Ca
2+ content
[125][139][125,139]. The MPTP opening leads to loss of mitochondrial inner membrane potential, oxidative phosphorylation/ATP production uncoupling, mitochondrial membrane rupture, release of apoptotic factors (cytochrome c) and cell death by necrosis
[36][124][36,124].
Blood cells contribute to reperfusion-related microvasculature injury and MVO. Serial CMR imaging studies in pigs have shown that interstitial edema is maximal immediately after reperfusion, whereas the maximal content of neutrophils, macrophages, and collagen is observed at 24 h, 4 days and 7 days after reperfusion
[135]. Neutrophils brought to ischemic microcirculation upon blood restoration are activated and tend to aggregate with other neutrophils, platelets or endothelial cells leading to microcirculation plugging and MVO. Activated neutrophils produce inflammatory cytokines, ROS, elastase and metalloproteinases, which cause capillary destruction, vascular leakage and a strong inflammatory response. Newly brought platelets are activated in the highly prothrombotic microcirculation undergoing ischemia/reperfusion, and activated platelets aggregate causing further capillary plugging and release numerous active substances with vasopressor and prothrombotic effects further increasing vascular tone and microthrombi formation (see: Myocardial ischemia). Thus, newly arrived cells in previously ischemic microcirculation are activated and recruited in capillaries and further accentuate ischemia-induced MVO and CNR.
Intramyocardial hemorrhage is one of the most severe manifestations of reperfusion-related injury that is closely linked with MVO and CNR
[140][141][140,141]. Experimental studies in anesthetized dogs
[142] and autopsy studies in patients with AMI (recanalized with intracoronary streptokinase within 3.5 h of ischemia)
[143] showed that intramyocardial hemorrhage is always observed following reperfusion but not in nonreperfused infarctions. Intramyocardial hemorrhage is always confined to the necrotic zone, predominantly in the central part of the necrosis, and tends to diminish towards the border zone
[143][144][143,144]. Serial imaging studies in pigs have shown that the hemorrhage score (from 0 absent to 5 very severe) was 0 at 120 min, 2 at 24 h, 4 at day 4 and 1 at day 7 after the reperfusion
[135]. CMR imaging and histological studies in dogs undergoing 4 h of coronary occlusion followed by 1 h of reperfusion showed that CMR-assessed hemorrhage size (decreased signal intensity zones) correlated closely with hemorrhage size defined by histology (correlation coefficient of 0.96)
[145]. In dogs without reperfusion, no macroscopic or CMR-defined zones of hemorrhage were observed
[145]. Marked increase in permeability and gap formation (of sufficient diameter to allow passage of erythrocytes) in the vascular barrier after ischemia followed by reperfusion may lead to extravasation of red blood cells in the perivascular space. Platelet-activating factor (PAF)—a potent inflammatory mediator
[146]—and prolonged adhesion of neutrophils to endothelium may promote gap formation
[147] via basal membrane destruction and endothelial cell detachment via released active proteases
[147] and formation of neutrophil extracellular traps (NETs)
[74][148][74,148]. Intramyocardial hemorrhage is more common after prolonged severe ischemia followed by reperfusion, which results in necrosis of endothelial cells, breakdown of basal membrane and destroyed microvessels
[149][150][149,150]. These studies support the notion that intramyocardial hemorrhage represents the most severe form of ischemia/reperfusion. Microvascular destruction and local consumption of coagulation factors due to coagulation cascade activation and intravascular microthrombi formation promoted by activated endothelium and inflammation have also been suggested as mechanisms for extravasation of erythrocytes and hemorrhage in areas of MVO after AMI
[151]. Apart from being a manifestation of severity of ischemia/reperfusion after AMI, intramyocardial hemorrhage per se aggravates MVO and CNR through extracellular compression and other mechanisms. CMR imaging studies in swine undergoing circumflex coronary artery occlusion for 75 min by a balloon catheter and in patients with AMI showed an overlap and a close anatomic correlation between areas of intramyocardial hemorrhage and MVO (correlation coefficients of 0.85 and 0.87, respectively)
[151]. In addition, CMR imaging studies showed that all patients with AMI and intramyocardial hemorrhage on T2* imaging had CMR-confirmed MVO
[152] or that 80% of patients with MVO had CMR evidence of intramyocardial hemorrhage
[153]. Intramyocardial hemorrhage is irreversible and induces a powerful and prolonged inflammatory response. Extravasated erythrocytes undergo destruction, which leads to iron release and deposition in the infarct zone. CMR imaging studies in canine models of ischemia/reperfusion showed that intramyocardial hemorrhage leads to iron deposition in the infarct zone up to 2 months after the acute event and that newly recruited macrophages colocalize with iron deposits, suggesting a prolonged inflammatory burden in the chronic phase of myocardial infarction
[154]. Intramyocardial hemorrhage appears to be more frequent after reperfusion by thrombolytic agents than primary angioplasty. In a series of 19 necropsies of patients undergoing thrombolytic therapy, 74% of infarcts treated with thrombolytic agents (but none of the infarcts undergoing balloon angioplasty alone) were hemorrhagic
[155]. Intramyocardial hemorrhage appears to be associated (or be more frequent) with larger infarct size, greater MVO, larger left ventricular dimensions, lower left ventricular ejection fraction (LVEF), anterior wall infarct location and glycoprotein IIb/IIIa inhibitor use. However, after adjustment, only anterior wall infarct location and the use of glycoprotein IIb/IIIa inhibitors were associated with higher odds of intramycardial hemorrhage
[156]. Intramyocardial hemorrhage is a determinant of infarct size, infarct expansion and reduced myocardial salvage after reperfusion
[157]. Intramyocardial hemorrhage is a strong correlate of adverse outcomes, which is stronger than infarct size
[152] and patients with MVO and intramyocardial hemorrhage have a worse prognosis than patients with MVO without intramyocardial hemorrhage
[158].
Ischemia/reperfusion is associated with a strong inflammatory response in the infarct zone, predominantly mediated by neutrophils, which contributes to MVO and CNR. Monocytes, macrophages and lymphocytes do participate in the inflammatory response, as well. One experimental study in mongrel dogs, in which a segment of a large epicardial coronary artery was deprived of blood flow for 3 h followed by reperfusion, showed an influx of neutrophils within the media of ischemic/reperfused vessels but not in the nonischemic vessels. Electron microscopic analysis showed that neutrophils were often located between the endothelial cells and the elastic lamina in the ischemic/reperfused vessels
[159]. The deleterious effects of neutrophils (and other immune cells) are predominantly mediated by release various active substances including inflammatory cytokines
[160], MMPs (particularly MMP-9)
[77][78][77,78], ROS
[75][98][75,98] and myeloperoxidase
[76]. The necrotic debris (among other stimuli) activates the NLRP3 inflammasome, which regulates caspase-1 activity, stimulates production (and release) of large amounts of cytokines (primarily IL-1β and IL-18) and promotes inflammatory cell death via pyroptosis
[161]. Inflammasome contributes to MVO and CNR by exacerbating endothelial cell damage and vascular leakage (promoting interstitial edema and microcirculation compression), heightening vasopressor tone and promoting cellular trapping and stasis and microthrombi formation in microcirculation
[162][163][162,163]. Patients with AMI who develop MVO and CNR have higher levels of several inflammatory cytokines in circulation, including C-reactive protein
[127][164][127,164], interleukin 6 (IL-6)
[165] and interleukin 8 (IL-8)
[166] compared with patients who did not develop these phenomena. Patients developing CNR have significantly higher levels of myeloperoxidase at culprit lesions than patients without CNR
[167][168][167,168]. Myeloperoxidase produces a large number of highly reactive species which attack all known cellular components leading to reduced NO availability, endothelial dysfunction and impaired vasoreactivity
[76].
Of all mechanisms and mediators proposed to date to explain microcirculation damage in the setting of ischemia/reperfusion, ROS and MMPs have received most attention for their role in the genesis of MVO. Excess production of ROS and activation (or overexpression) of MMPs are underlying mechanisms of tissue damage (including microcirculation) during ischemia/reperfusion and their actions appear to be mutually dependent. In the setting of ischemia/reperfusion, ROS appear to have multiple cellular sources (endothelial cells, platelets, neutrophils or other immune cells and resident macrophages) and the main producers at the molecular level are xanthine oxidase, NADPH oxidase, mitochondrial electron transport chain and uncoupled NO synthase
[54][98][54,98]. Excess amounts of ROS interact with any biological structure in their vicinity, rendering them dysfunctional. ROS are involved and play a critical role in almost all cellular and molecular events leading to MVO in the setting of ischemia/reperfusion. Activated MMPs appear to have multiple sources including endothelial cells, smooth muscle cells, inflammatory cells and resident macrophages
[54]. MMPs have a wide specificity and cleave a wide range of extracellular matrix components including glycocalyx, inter-endothelial cell junctions, basal membrane and extracellular substrates such as adhesion molecules, cytokines and chemokines
[54]. MMPs play a critical role in the MVO by participation in the increased vascular permeability and leakage, glycocalyx shedding, capillary destruction and intramyocardial hemorrhage. Detailed information on the biology of ROS and MMPs and their role in genesis of MVO during ischemia/reperfusion are provided in 2 excellent reviews by Granger and Kvietys
[54][98][54,98].