Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wu, Q. Atrial Natriuretic Peptide Signaling in Uteroplacental Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/47928 (accessed on 24 July 2026).
Wu Q. Atrial Natriuretic Peptide Signaling in Uteroplacental Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/47928. Accessed July 24, 2026.
Wu, Qingyu. "Atrial Natriuretic Peptide Signaling in Uteroplacental Cells" Encyclopedia, https://encyclopedia.pub/entry/47928 (accessed July 24, 2026).
Wu, Q. (2023, August 11). Atrial Natriuretic Peptide Signaling in Uteroplacental Cells. In Encyclopedia. https://encyclopedia.pub/entry/47928
Wu, Qingyu. "Atrial Natriuretic Peptide Signaling in Uteroplacental Cells." Encyclopedia. Web. 11 August, 2023.
Copy Citation
Endometrial decidualization is a uterine process essential for spiral artery remodeling, embryo implantation, and trophoblast invasion. Defects in endometrial decidualization and spiral artery remodeling are important contributing factors in preeclampsia, a major disorder in pregnancy. Atrial natriuretic peptide (ANP) is a cardiac hormone that regulates blood volume and pressure. ANP is also generated in non-cardiac tissues, such as the uterus and placenta. In recent human genome-wide association studies, multiple loci with genes involved in natriuretic peptide signaling are associated with gestational hypertension and preeclampsia.
ANP
corin
endometrial decidualization
natriuretic peptides
1. Introduction
Pregnancy is associated with major hormonal changes, including increased production and release of chorionic gonadotropin from trophoblasts and progesterone from the corpus luteum in the ovary. These hormones induce profound cellular and functional changes in the uterine tissue, e.g., endometrial decidualization and spiral artery remodeling. Such changes are essential for embryo implantation and an adequate uteroplacental blood flow to support the developing placenta and fetus.
Preeclampsia is a disorder characterized by the new onset of high blood pressure after ~20 weeks of pregnancy [1][2]. It occurs in ~4–8% of all pregnancies and often leads to damages in major organs, including the kidney, liver, and placenta [3]. The cause of preeclampsia is not fully understood. One of the common pathological findings in the preeclamptic uterus is incompletely remodeled spiral arteries, which limits blood flow to the placenta [4][5]. Placental ischemia and oxidative stress in turn lead to the release of detrimental placental factors into the maternal circulation, causing inflammatory responses, endothelial dysfunction, and systemic complications [4][6]. It has been shown that aberrant expression and/or function of angiogenic factors, e.g., vascular endothelial growth factor (VEGF) and its receptors, e.g., soluble VEGF receptor 1, also called soluble fms-like tyrosine kinase-1 (sFlt1), play a major role in the pathogenesis of preeclampsia [1][7].
Poor endometrial decidualization, also called decidualization resistance, is another pathological characteristic in preeclampsia [8][9][10]. Decreased circulating levels of insulin-like growth factor-binding protein-1 (IGFBP-1), an endometrial decidualization marker, in preeclamptic women are well documented [9][11]. These findings indicate that impaired molecular mechanisms controlling uterine endometrial decidualization and tissue remodeling are potential contributing factors in preeclampsia.
Atrial natriuretic peptide (ANP) is a versatile hormone produced largely in cardiomyocytes [12][13][14]. The main function of ANP is to control electrolyte homeostasis, body fluid balance, and blood pressure. Studies in cultured cells and mouse models indicate that ANP also acts in the pregnant uterus to stimulate endometrial decidualization, spiral artery remodeling, and trophoblast invasion [15][16][17]. Recent large-scale genetic studies in humans have also implicated impaired ANP generation and signaling in gestational hypertension and preeclampsia [18][19].
2. The Soil, Seeds, and Roots
Endometrial decidualization involves a cascade of gene expression and cellular changes necessary for embryo implantation and trophoblast invasion [20]. Trophoblast invasion is a process, in which trophoblast cells from the developing embryo invade the decidualized endometrium and make connections with maternal spiral arteries [21][22]. This process is crucial for the access of an adequate maternal blood supply to nourish the developing fetus.
In an analogy of soil and seed, the decidualized endometrium represents the receptive soil that provides a fertile environment for invading trophoblasts, i.e., the seeds. For the trophoblasts to grow effectively, endometrial decidualization must be adequate. Upon entering the decidualized endometrium, the trophoblasts interact with the decidual cells, extend invasive projections, like roots, into the decidualized tissue, and release signaling molecules to modify the endometrium, making it easier for the roots to extend. Such an intricate interplay between the decidual cells and the invading trophoblasts is critical for proper spiral artery remodeling, a healthy maternal–fetal interface, and an ultimately successful pregnancy.
Poor spiral artery remodeling, reduced uteroplacental blood flow, and ensuing placental ischemia have been recognized as central pathological mechanisms in preeclampsia. Increased systemic blood pressure in preeclamptic women may reflect, in part, a compensatory response to placental ischemic signals, to maintain steady blood flow in the presence of narrow uterine spiral arteries. Endometrial decidualization is essential for spiral artery remodeling [20][23]. Thus, poorly decidualized endometrium could be a major reason for defective spiral artery remodeling in preeclampsia. Indeed, studies with uterine samples from preeclamptic women have shown altered gene and protein profiles that are indicative of improper endometrial decidualization and sFlt1 expression [10][24]. Decreased circulating levels of decidualization markers, such as prolactin and IGFBP-1, have also been found in preeclamptic women [11][25]. These findings support the idea that defective endometrial decidualization is an important mechanism in the pathogenesis of preeclampsia. These studies also point to ANP signaling as a potential regulatory mechanism in early cellular events of decidualization and spiral artery remodeling, as well as in subsequent trophoblast invasion in the pregnant uterus.
3. Role of ANP in Decidual and Vascular Cell Interactions
3.1. Decidualization and Spiral Artery Remodeling
Endometrial decidualization and uterine spiral artery remodeling occur before direct contact with invading trophoblasts [26]. In fact, morphological changes in endometrial arteries were observed in women with ectopic pregnancies, i.e., the implantation occurs outside the uterus [26]. These findings indicate that the initial cellular events in spiral artery remodeling are mediated mostly by signaling molecules from decidual stromal cells, but not trophoblasts.
Studies in animal models also support the role of decidual stromal cells in regulating cellular changes in spiral arteries. In mice, for example, apoptosis is markedly increased in the pregnant uterus [15]. Sequential death in smooth muscle cells and endothelial cells was observed in endometrial arterial segments, where no adjacent intravascular or interstitial trophoblast invasion was detected [15]. In cell culture, decidualized human endometrial stromal cells were found to secrete TNF-related apoptosis-inducing ligand (TRAIL), a death protein that triggered apoptosis in uterine smooth muscle cells [15]. Subsequently, the apoptotic smooth muscle cells released cyclophilin B, a proinflammatory protein that upregulates TRAIL receptor in cancer cells via MAPK/ERK signaling [27][28]. Similar cyclophilin B-mediated MAPK/ERK signaling and TRAIL receptor upregulation were observed in cultured uterine endothelial cells. As a result, the uterine endothelial cells became responsive to TRAIL-induced cell death [15]. These findings could explain the observed sequential cell death, first in arterial smooth muscle cells and then in endothelial cells in the mouse pregnant uterus.
In pregnant ANP KO mice, uterine endometrial decidualization is compromised, as indicated by reduced prolactin and IGFBP-1 expression [15]. When cultured human endometrial stromal cells were treated with ANP, prolactin, IGFBP-1, and TRAIL expression was increased. The conditioned medium from the ANP-treated endometrial stromal cells also had an increased activity in inducing apoptosis in uterine smooth muscle cells [15]. On the other hand, ANP treatment in cultured human endothelial cells did not alter cell proliferation and migration or prevent TNFα-induced dysfunction [29]. These findings are consistent, indicating an important role for ANP in inducing TRAIL expression and release from endometrial stromal cells, causing apoptosis in arterial smooth muscle cells [15]. Cyclophilin B release from smooth muscle cells and TRAIL receptor induction eventually lead to apoptosis in arterial endothelial cells. These well-orchestrated cellular events are probably an important part of the spiral artery remodeling process. The loss of arterial smooth muscle cells and endothelial cells may pave the way for intravascular invasion of placental trophoblasts. More studies are required to understand the ANP-mediated signaling mechanism in uterine stromal cells.
ANP is expressed in the uterus and placenta [17][30][31]. In principle, the effects on decidual stromal cells could be due to ANP from either the uterus or the placenta, or both. To distinguish the role of uterine ANP from placental ANP, mouse models with either uterine or placental ANP deficiency were analyzed [15]. As indicated by reduced prolactin and IGFBP1 expression, uterine decidualization was defective in mice lacking either uterine or placental ANP, indicating that ANP from both tissues contributes to the decidualization process [15]. However, impaired decidualization was more profound in mice lacking uterine ANP than placental ANP, indicating that ANP made in the uterus is most critical for decidualization. Consistently, studies in Corin KO mice also show that uterine corin is more important than placental corin in promoting spiral artery remodeling [16]. In addition, low uterine TRAIL levels were observed in pregnant ANP and Corin KO mice [15], which exhibit a preeclampsia-like phenotype. Thus, in addition to its vasorelaxation function, uterine ANP, activated by corin in situ, may promote decidualization and TRAIL expression in endometrial stromal cells. The stromal cell-derived TRAIL induces arterial smooth muscle cell death and cyclophilin B release. Cyclophilin B in turn upregulates TRAIL receptor expression in endothelial cells, subjecting the cells to TRAIL-induced apoptosis (Figure 1). Defects in the corin and ANP function are expected to impair endometrial decidualization and spiral artery remodeling, thereby contributing to gestational hypertension and preeclampsia. Additional investigations will be important to verify this hypothesis in other animal models and to test if a similar mechanism exists in the human pregnant uterus. It will be also important to examine if ANP signaling contributes to uterine structural changes during the normal menstrual cycle in humans.
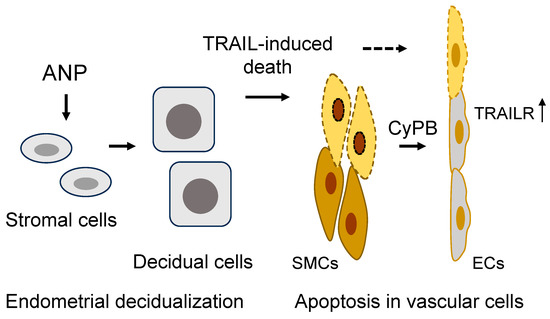
Figure 1. A model of ANP function in endometrial decidualization and spiral artery remodeling in the pregnant uterus. In the pregnant uterus, ANP stimulates stromal cell decidualization and TNF-related apoptosis-inducing ligand (TRAIL) expression and secretion, inducing apoptosis in arterial smooth muscle cells (SMCs). Apoptotic SMCs release cyclophilin B (CyPB) to upregulate TRAIL receptor (TRAILR) expression in endothelial cells (ECs), making the cells susceptible to TRAIL-mediated apoptosis. The sequential death in SMCs and ECs facilitates spiral artery remodeling.
3.2. Trophoblast Invasion
As discussed above, the initiation of endometrial decidualization and spiral artery remodeling is independent of trophoblasts [20]. However, embryo implantation and the subsequent trophoblast invasion are expected to act in a reciprocal manner to enhance endometrial decidualization. It is well documented that trophoblasts can secrete a variety of proteins, such as proteolytic enzymes, cytokines, and signaling molecules, to promote endometrial decidualization, extracellular matrix degradation, and uterine tissue remodeling [32][33][34]. Trophoblasts also secrete proapoptotic factors, such as TRAIL, TNFα, and FAS ligand, to induce apoptosis in arterial smooth muscle cells and endothelial cells, as shown in cell experiments and mouse models [35]. These activities are expected to accelerate the structural changes in arterial walls and facilitate trophoblast penetration, leading to the transformation of the uterine arteries from high- to low-resistance vessels. Such transformation ensures an increased uteroplacental blood flow with low velocity, which is necessary for supporting the growth of the developing fetus [36].
The placenta is another site for corin and pro-ANP expression, indicating a role of ANP in trophoblast function [15][37][38]. In supporting this hypothesis, the ANP receptor NPR-A is expressed in human trophoblasts and decidual stromal cells [16][39]. In cultured human trophoblasts, ANP treatment increases intracellular cGMP levels and promotes trophoblast invasion in Matrigel assays, whereas NPR-A knockdown suppresses trophoblast invasion [16][40][41]. Moreover, markedly decreased placental corin and NPR-A expression and increased levels of pro-ANP are found in preeclamptic women [40][41]. These results indicate that ANP-mediated signaling probably has a role in promoting trophoblast invasion, and that defects in ANP signaling may contribute to uterine and/or placental pathology in preeclampsia.
The ANP signaling mechanism in trophoblasts is not fully understood. In the heart, ANP modulates autophagy, a process of regulating cellular function depending on pathophysiological conditions [42][43]. In a recent study in human trophoblasts and chorionic villi, a molecular mechanism has been suggested, in which ANP activates NPR-A and subsequent adenosine 5′-monophosphate-activated protein kinase (AMPK) and the mammalian target of rapamycin complex 1 (mTORC1) signaling to inhibit autophagy and induce protein kinase N3 (PKN3) expression, thereby increasing metalloproteinase expression and trophoblast invasiveness [41] (Figure 2). Autophagy activation or downregulation of PKN3 diminishes the effect of ANP on trophoblast invasion [41]. Moreover, placentas from preeclamptic women had reduced PKN3 protein levels [41]. These results are intriguing, indicating a potential link between impaired ANP signaling and low PKN3 activity in the pathogenesis of preeclampsia. It appears that ANP can stimulate multiple signaling pathways to modify a wide range of cellular functions in endometrial decidualization and trophoblast invasion, which are important in pregnancy.
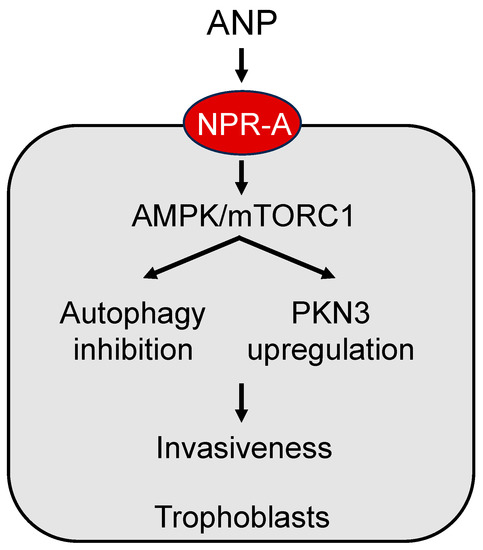
Figure 2. A model of ANP signaling in placental trophoblasts. ANP activates natriuretic peptide receptor A (NPR-A) to enhance adenosine 5′-monophosphate-activated protein kinase (AMPK) and the mammalian target of rapamycin complex 1 (mTORC1) signaling, leading to autophagy inhibition and protein kinase N3 (PKN3) upregulation, thereby increasing trophoblast invasiveness.
References
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; Whitehead, C.; Hyett, J.; da Silva Costa, F.; Nicolaides, K.; et al. Pre-eclampsia. Nat. Rev. Dis. Primers 2023, 9, 8.
- Magee, L.A.; Nicolaides, K.H.; von Dadelszen, P. Preeclampsia. N. Engl. J. Med. 2022, 386, 1817–1832.
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7.
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381.
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594.
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480.
- Massri, N.; Loia, R.; Sones, J.L.; Arora, R.; Douglas, N.C. Vascular changes in the cycling and early pregnant uterus. JCI Insight 2023, 8, e163422.
- Garrido-Gómez, T.; Castillo-Marco, N.; Cordero, T.; Simón, C. Decidualization resistance in the origin of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S886–S894.
- Conrad, K.P.; Rabaglino, M.B.; Post Uiterweer, E.D. Emerging role for dysregulated decidualization in the genesis of preeclampsia. Placenta 2017, 60, 119–129.
- Garrido-Gomez, T.; Dominguez, F.; Quiñonero, A.; Diaz-Gimeno, P.; Kapidzic, M.; Gormley, M.; Ona, K.; Padilla-Iserte, P.; McMaster, M.; Genbacev, O.; et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc. Natl. Acad. Sci. USA 2017, 114, E8468–E8477.
- Liao, S.; Vickers, M.H.; Taylor, R.S.; Jones, B.; Fraser, M.; McCowan, L.M.E.; Baker, P.N.; Perry, J.K. Maternal serum IGF-1, IGFBP-1 and 3, and placental growth hormone at 20weeks’ gestation in pregnancies complicated by preeclampsia. Pregnancy Hypertens. 2017, 10, 149–154.
- Song, W.; Wang, H.; Wu, Q. Atrial natriuretic peptide in cardiovascular biology and disease (NPPA). Gene 2015, 569, 1–6.
- Rao, S.; Pena, C.; Shurmur, S.; Nugent, K. Atrial Natriuretic Peptide: Structure, Function, and Physiological Effects: A Narrative Review. Curr. Cardiol. Rev. 2021, 17, e051121191003.
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717.
- Zhang, W.; Li, S.; Lou, J.; Li, H.; Liu, M.; Dong, N.; Wu, Q. Atrial natriuretic peptide promotes uterine decidualization and a TRAIL-dependent mechanism in spiral artery remodeling. J. Clin. Investig. 2021, 131, e151053.
- Cui, Y.; Wang, W.; Dong, N.; Lou, J.; Srinivasan, D.K.; Cheng, W.; Huang, X.; Liu, M.; Fang, C.; Peng, J.; et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature 2012, 484, 246–250.
- Zhou, Y.; Wu, Q. Role of corin and atrial natriuretic peptide in preeclampsia. Placenta 2013, 34, 89–94.
- Honigberg, M.C.; Truong, B.; Khan, R.R.; Xiao, B.; Bhatta, L.; Vy, H.M.T.; Guerrero, R.F.; Schuermans, A.; Selvaraj, M.S.; Patel, A.P.; et al. Polygenic prediction of preeclampsia and gestational hypertension. Nat. Med. 2023, 9, 1540–1549.
- Tyrmi, J.S.; Kaartokallio, T.; Lokki, A.I.; Jääskeläinen, T.; Kortelainen, E.; Ruotsalainen, S.; Karjalainen, J.; Ripatti, S.; Kivioja, A.; Laisk, T.; et al. Genetic Risk Factors Associated with Preeclampsia and Hypertensive Disorders of Pregnancy. JAMA Cardiol. 2023, 8, 674–683.
- Gellersen, B.; Brosens, J.J. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr. Rev. 2014, 35, 851–905.
- Pijnenborg, R.; Robertson, W.B.; Brosens, I.; Dixon, G. Review article: Trophoblast invasion and the establishment of haemochorial placentation in man and laboratory animals. Placenta 1981, 2, 71–91.
- Knöfler, M.; Pollheimer, J. Human placental trophoblast invasion and differentiation: A particular focus on Wnt signaling. Front. Genet. 2013, 4, 190.
- Sharma, S.; Godbole, G.; Modi, D. Decidual Control of Trophoblast Invasion. Am. J. Reprod. Immunol. 2016, 75, 341–350.
- Sahu, M.B.; Deepak, V.; Gonzales, S.K.; Rimawi, B.; Watkins, K.K.; Smith, A.K.; Badell, M.L.; Sidell, N.; Rajakumar, A. Decidual cells from women with preeclampsia exhibit inadequate decidualization and reduced sFlt1 suppression. Pregnancy Hypertens. 2019, 15, 64–71.
- Yuen, B.H.; Cannon, W.; Woolley, S.; Charles, E. Maternal plasma and amniotic fluid prolactin levels in normal and hypertensive pregnancy. Br. J. Obstet. Gynaecol. 1978, 85, 293–298.
- Craven, C.M.; Morgan, T.; Ward, K. Decidual spiral artery remodelling begins before cellular interaction with cytotrophoblasts. Placenta 1998, 19, 241–252.
- Liao, D.F.; Jin, Z.G.; Baas, A.S.; Daum, G.; Gygi, S.P.; Aebersold, R.; Berk, B.C. Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 189–196.
- Kim, K.; Kim, H.; Jeong, K.; Jung, M.H.; Hahn, B.S.; Yoon, K.S.; Jin, B.K.; Jahng, G.H.; Kang, I.; Ha, J.; et al. Release of overexpressed CypB activates ERK signaling through CD147 binding for hepatoma cell resistance to oxidative stress. Apoptosis 2012, 17, 784–796.
- Binder, N.K.; Beard, S.; de Alwis, N.; Fato, B.R.; Nguyen, T.V.; Kaitu′u-Lino, T.J.; Hannan, N.J. Investigating the Effects of Atrial Natriuretic Peptide on the Maternal Endothelium to Determine Potential Implications for Preeclampsia. Int. J. Mol. Sci. 2023, 24, 6182.
- Reis, A.M.; Jankowski, M.; Mukaddam-Daher, S.; Tremblay, J.; Dam, T.V.; Gutkowska, J. Regulation of the natriuretic peptide system in rat uterus during the estrous cycle. J. Endocrinol. 1997, 153, 345–355.
- Graham, C.H.; Watson, J.D.; Blumenfeld, A.J.; Pang, S.C. Expression of atrial natriuretic peptide by third-trimester placental cytotrophoblasts in women. Biol. Reprod. 1996, 54, 834–840.
- Staun-Ram, E.; Shalev, E. Human trophoblast function during the implantation process. Reprod. Biol. Endocrinol. 2005, 3, 56.
- Liu, Y.; Fan, X.; Wang, R.; Lu, X.; Dang, Y.L.; Wang, H.; Lin, H.Y.; Zhu, C.; Ge, H.; Cross, J.C.; et al. Single-cell RNA-seq reveals the diversity of trophoblast subtypes and patterns of differentiation in the human placenta. Cell Res. 2018, 28, 819–832.
- Marsh, B.; Blelloch, R. Single nuclei RNA-seq of mouse placental labyrinth development. eLife 2020, 9, e60266.
- Whitley, G.S.; Cartwright, J.E. Trophoblast-mediated spiral artery remodelling: A role for apoptosis. J. Anat. 2009, 215, 21–26.
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482.
- Abassi, Z.; Kinaneh, S.; Skarzinski, G.; Cinnamon, E.; Smith, Y.; Bursztyn, M.; Ariel, I. Aberrant corin and PCSK6 in placentas of the maternal hyperinsulinemia IUGR rat model. Pregnancy Hypertens. 2020, 21, 70–76.
- Degrelle, S.A.; Chissey, A.; Stepanian, A.; Fournier, T.; Guibourdenche, J.; Mandelbrot, L.; Tsatsaris, V. Placental Overexpression of Soluble CORIN in Preeclampsia. Am. J. Pathol. 2020, 190, 970–976.
- Gerbes, A.L.; Dagnino, L.; Nguyen, T.; Nemer, M. Transcription of brain natriuretic peptide and atrial natriuretic peptide genes in human tissues. J. Clin. Endocrinol. Metab. 1994, 78, 1307–1311.
- Tan, H.; Lin, L.; Huang, L.; Yu, Y. Is Atrial Natriuretic Peptide (ANP) and Natriuretic Peptide Receptor-A (NPR-A) Expression in Human Placenta and Decidua Normal? Med. Sci. Monit. 2019, 25, 2868–2878.
- Chu, N.; Tang, Y.; Wang, C.J.; Pei, J.N.; Luo, S.L.; Yu, Y.; Liu, Z.Z.; Liu, H.Y.; Qiu, X.M.; Wang, L.; et al. ANP promotes HTR-8/SVneo cell invasion by upregulating protein kinase N 3 via autophagy inhibition. FASEB J. 2023, 37, e22779.
- Forte, M.; Marchitti, S.; Di Nonno, F.; Stanzione, R.; Schirone, L.; Cotugno, M.; Bianchi, F.; Schiavon, S.; Raffa, S.; Ranieri, D.; et al. NPPA/atrial natriuretic peptide is an extracellular modulator of autophagy in the heart. Autophagy 2023, 19, 1087–1099.
- Raffa, S.; Forte, M.; Gallo, G.; Ranieri, D.; Marchitti, S.; Magrì, D.; Testa, M.; Stanzione, R.; Bianchi, F.; Cotugno, M.; et al. Atrial natriuretic peptide stimulates autophagy/mitophagy and improves mitochondrial function in chronic heart failure. Cell. Mol. Life Sci. 2023, 80, 134.
More
Information
Subjects:
Physiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
830
Revisions:
2 times
(View History)
Update Date:
11 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No