 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tapan Mukherjee | -- | 6461 | 2023-08-01 18:36:39 | | | |
| 2 | Peter Tang | Meta information modification | 6461 | 2023-08-02 07:48:41 | | |
Video Upload Options
Breast cancer (BC) currently occupies the second rank in cancer-related global female deaths. Although consistent awareness and improved diagnosis have reduced mortality, late diagnosis and resistant response still limit the therapeutic efficacy of chemotherapeutic drugs (CDs), leading to relapse with consequent invasion and metastasis. Treatment with CDs is indeed well-versed but it is badly curtailed with accompanying side effects and inadequacies of site-specific drug delivery. As a result, drug carriers ensuring stealth delivery and sustained drug release with improved pharmacokinetics and biodistribution are urgently needed. Core–shell mesoporous silica nanoparticles (MSNPs) have been a cornerstone in this context, attributed to their high surface area, low density, robust functionalization, high drug loading capacity, size–shape-controlled functioning, and homogeneous shell architecture, enabling stealth drug delivery.
1. Introduction
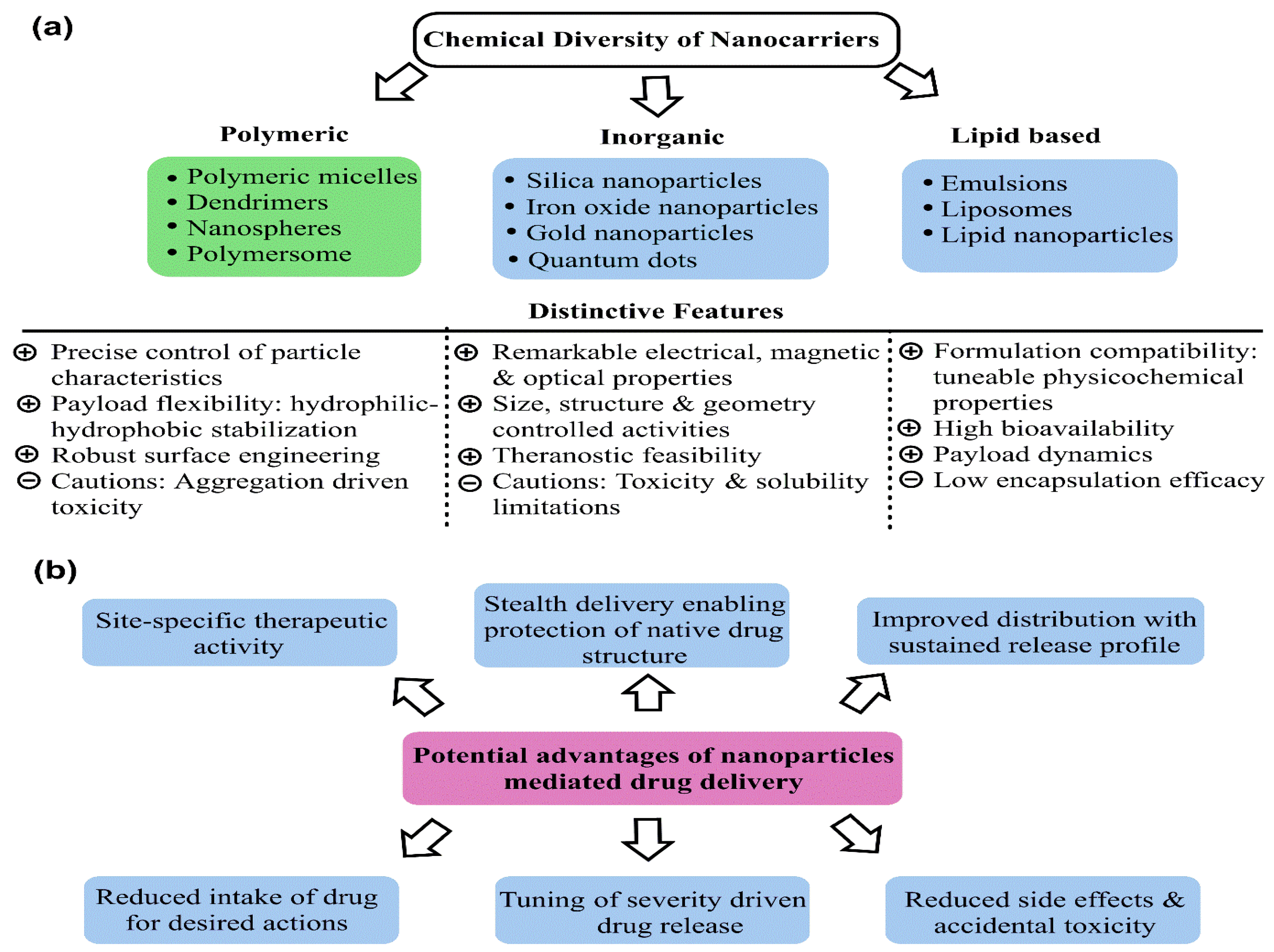
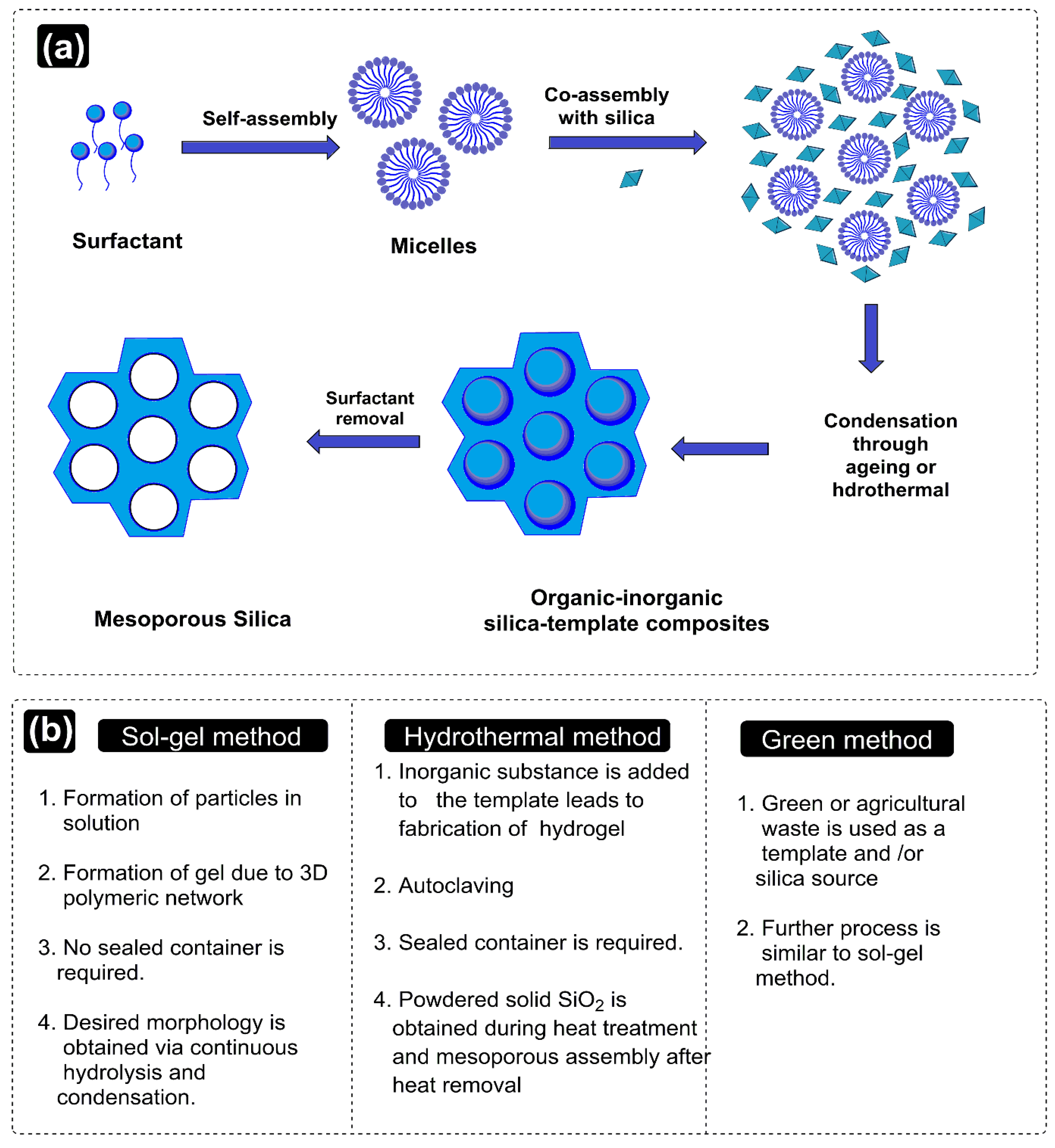
2. Exclusive Terminologies and Synthesis Methods of Mesoporous Silica Nanoparticles
2.1. Synthesis Methods
3. Promising Aspects of Mesoporous Silica Nanoparticles as Drug Vehicles
4. Drug Release Mechanisms of Mesoporous Silica Nanoparticles
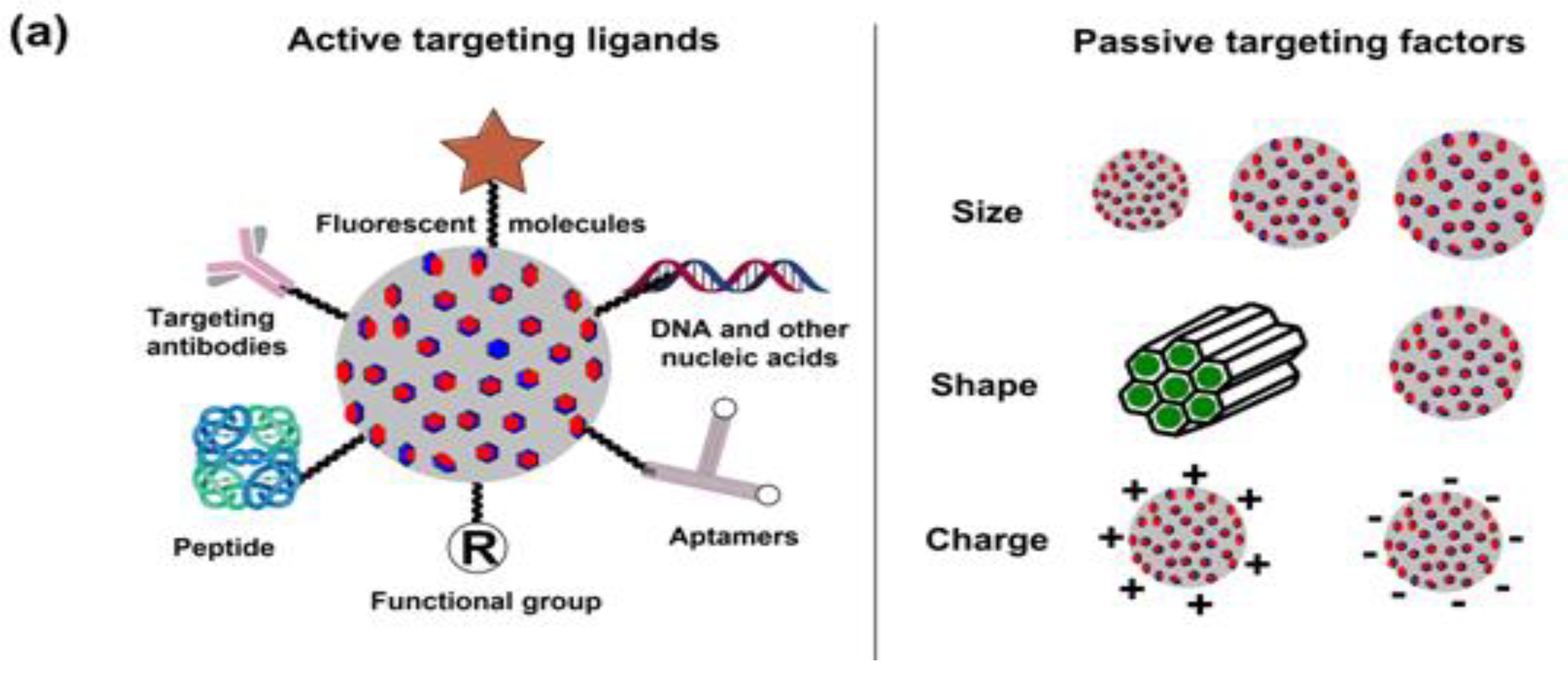
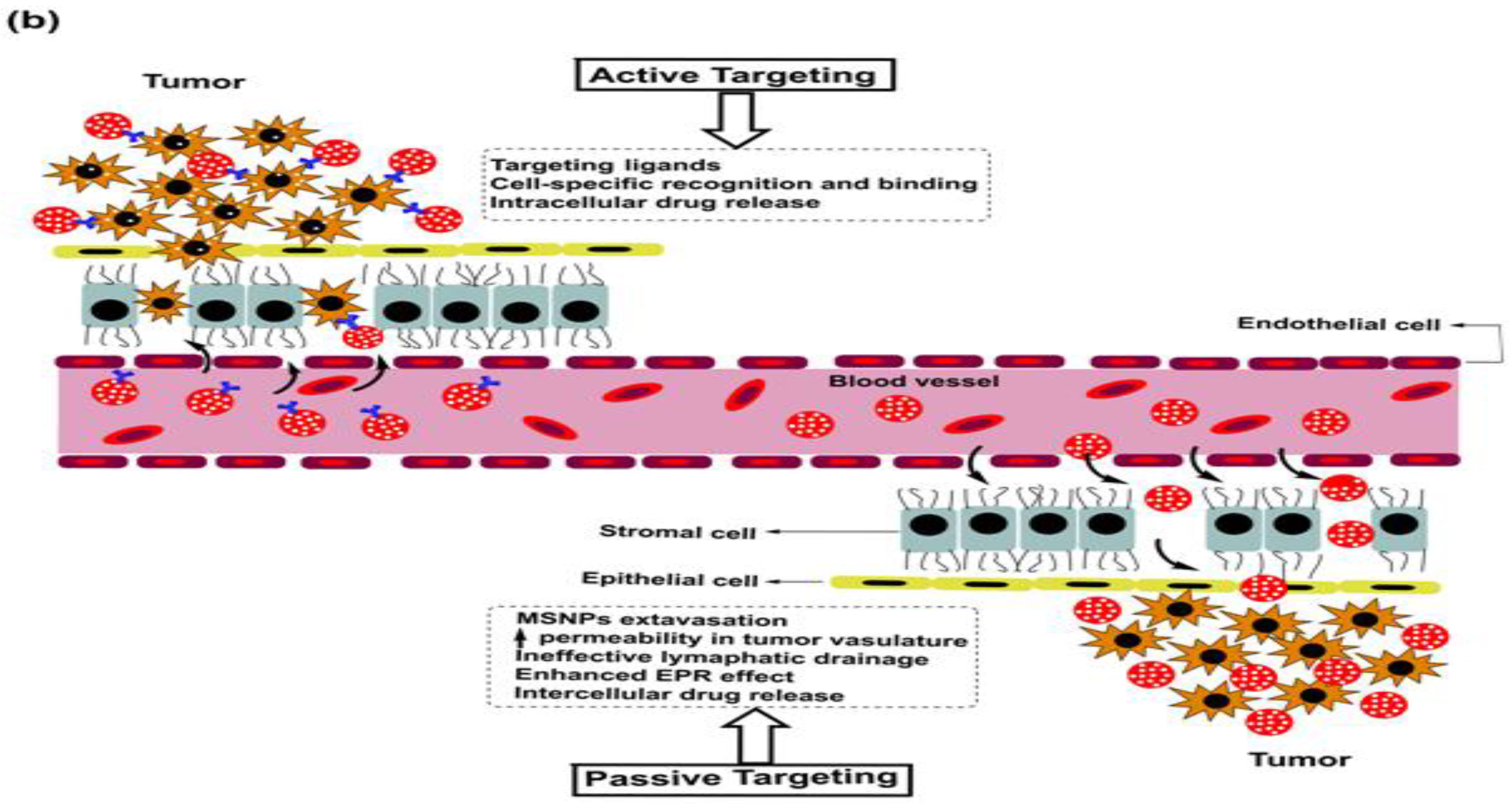
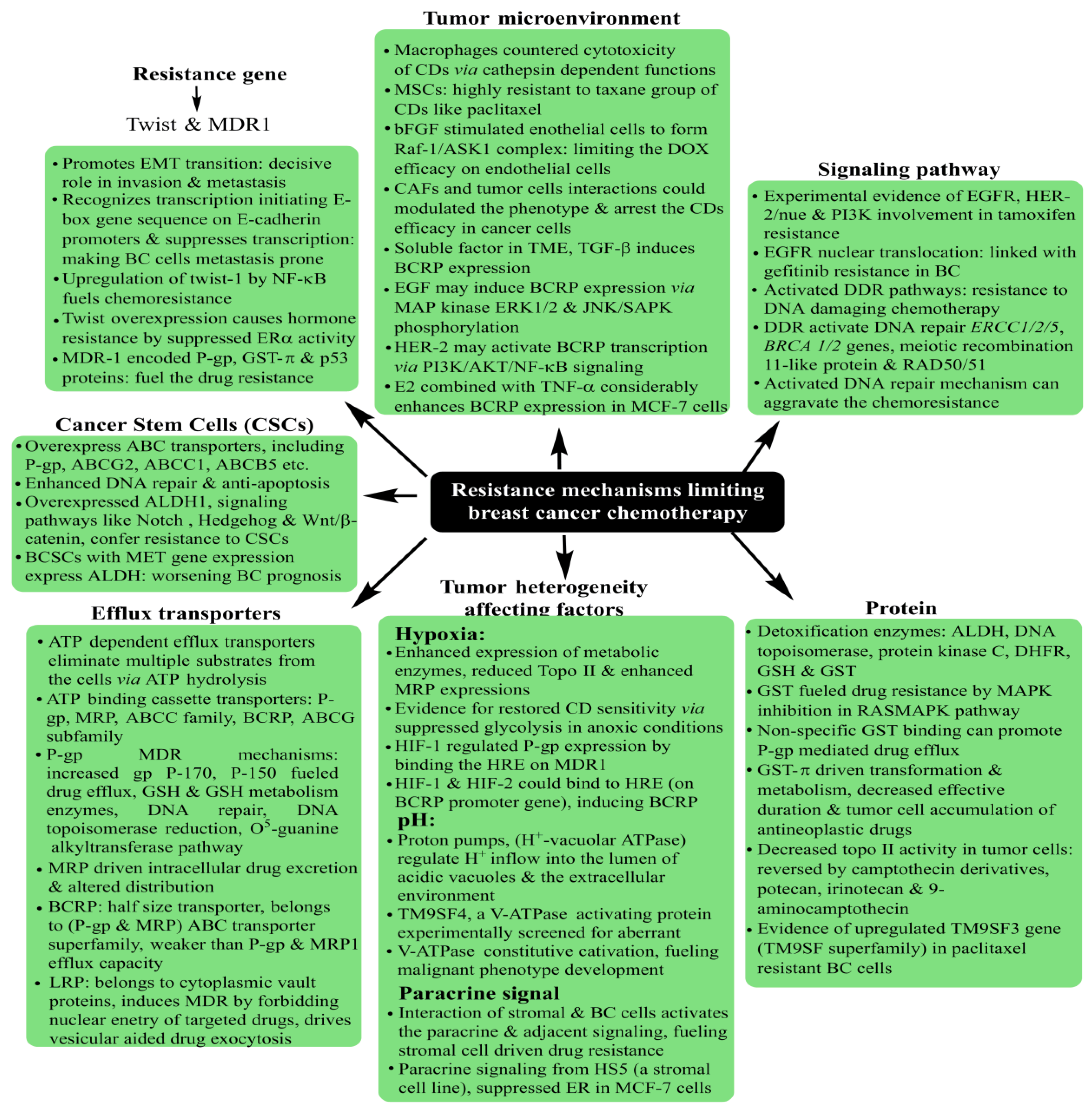
5. Breast Cancer: Pathophysiology, Mortality and Chemoresistance
|
Name of Receptor |
Physiological Distinctions |
Potential in Breast Cancer Treatment |
|---|---|---|
|
ErbB tyrosine kinase receptors |
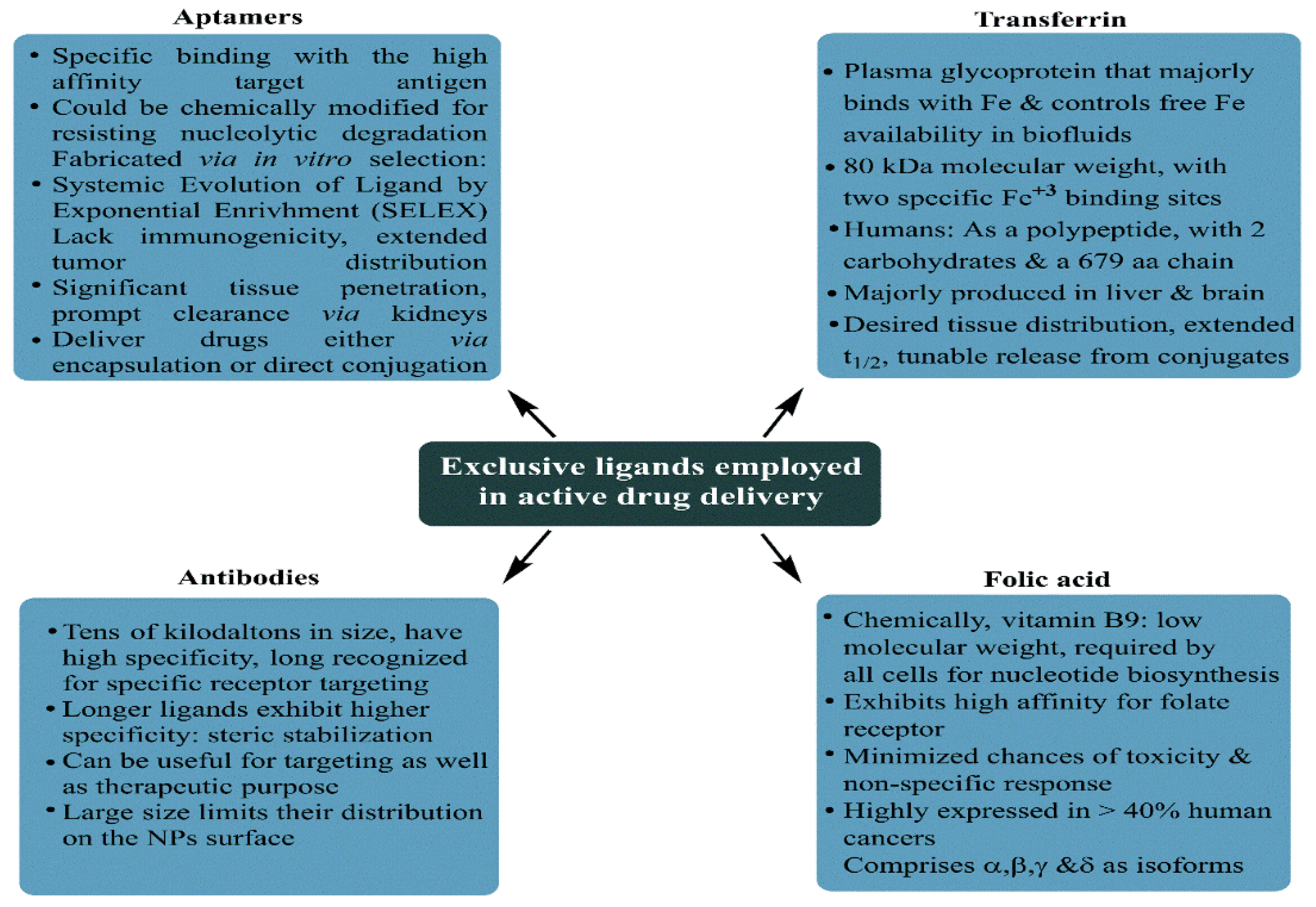
Widely investigated growth factor receptors in BC that consist of four homolog receptors: ErbB1 (HER1/EGFR), ErbB2 (HER2/neu), ErbB3 (HER3) and ErbB4 (HER4). EGFR and HER2 are overexpressed in 15–20% and 20–25% of BC cells. The BC cells overexpressing ErbB receptors demonstrate aggressive clinical behavior. |
Enhanced cytotoxicity of CURC delivered via EGFR targeting GE11 peptide-conjugated CURC-loaded PLGA-PEG NPs (in MCF-7 cells). HER2 Ab-conjugated PTX and rapamycin-loaded glycerol monooleate-coated magnetic NPs exhibited enhanced uptake in MCF-BC cells with 24-fold reduced toxicity and 3-fold lower IC50 extent (for PTX). For rapamycin, 71-fold reduced IC50 than anative drug with targeted magnetic NPs. EGFR-conjugated immuno NPs exhibited ~13-fold higher uptake and antiproliferative activity than unconjugated NPs in MCF-7 BC cells. HER2-targeted PLGA/montmorillonite-trastuzumab NPs exhibited 12.74 and 13.11-fold larger therapeutic efficacy than untargeted NPs. |
|
Folate receptor |
Binds to water-soluble, low-molecular-weight FA (vitamin B9), essential in normal mitotic cell division;, binds to the folate ligand and then internalizes the complexes via receptor-mediated endocytosis. |
Studied for high anticancer efficacy of PTX (in 4T1 BC cells), cisplatin (DDP) and docetaxel (in MDA-MB-231 BC cells), CURC (in MCF-7 cells), enhanced cellular uptake for deoxycholic acid-O-carboxymethylated CTS and vincristine sulfate-loaded PLGA-PEG NPs |
|
Estrogen receptors |
Belong to thenuclear hormone receptor superfamily, differentially overexpressed in 60–80% of BC cells, internalized in the cancer cells upon binding to ERs |
Enhanced cellular uptake of PEG-conjugated PTX-epirubicin co-loaded liposomal NPs, TAM surface-grafted liposomes loaded with DOX (in MCF-7 cells), DOX and DOX + TAM-loaded liposomes (MCF-7 cells) |
|
CD44/Hyaluronan receptor |
A natural component of ECM, hyaluronan is critically involved in cell proliferation, migration and invasion. CD44 regulates lymphocyte adhesion to endothelial cells during lymphocyte migration (a process equivalent to metastasis in solid tumors). CD44 prevails as an HA receptorandprevails sparsely on the surface of epithelial, hematopoietic and neuronal cells. Recent studies have screened CD44 as a BC stem cell marker. |
HA-functionalized CTS lipoic acid NPs loaded with 17α-methyltestosterone exhibited enhanced internalization in CD44 overexpressing BT-20 BC cells than CD44-negative MCF-7 cells. Enhanced uptake and cytotoxicity of PLGA NPs in MDA-MB-231 cells. Higher docetaxel uptake for self-assembled PLGA-HA block copolymers (in MDA-MB-231 cells) than untargeted NPs. HA matrix NPs with intrinsic CD-44 tropism and loaded rapamycin exhibited a 3.2-fold enhanced uptake in CD44-positive MDA-MB-468 cells. HA-lysine-lipoic acid NPs loaded with DOX exhibited a 20-fold enhanced uptake in MCF-7/ADR cells. |
|
Luteinizing hormone-releasing hormone receptor (LHRH) |
A hormonal decapeptide produced by the hypothalamus, LHRH is also known as a gonadotropin-releasing hormone. Plays a key role in regulating the pituitary–gonadal axis and reproduction. Overexpressed in 50% of BC cells. |
LHRH-conjugated PTX showed enhanced specificity for targeting MDA-MB-231 cells. LHRH-CTS-conjugated NPs enhanced cellular uptake with two-fold reduced IC50in LHR overexpressing MCF-7 cells than non-targeted NPs. DDP-loaded LHRH-modified dextran NPs exhibited a higher uptake with reduced nephrotoxicity of DDP than non-targeted NPs (in 4T1 BC cells). |
6. Discussion of Recent Attempts Using Mesoporous Silica Nanoparticles for Breast Cancer Treatment
6.1. Singular Drug Delivery Using Mesoporous Silica Nanoparticles
6.2. Combinatorial Drug Delivery Using Mesoporous Silica Nanoparticles
References
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66.
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.C.; Martin, M.; Namer, M.; et al. The global breast cancer burden: Variations in epidemiology and survival. Clin. Breast Cancer 2005, 6, 391–401.
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434.
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800.
- Muley, H.; Fad, R.; Rodriguez-Rodriguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Phamacol. 2020, 177, 113959.
- Lainetti, P.d.F.; Leis-Filho, A.F.; Laufer-Amorim, R.; Battazza, A.; Fonseca-Alves, C.E. Mechanisms of resistance to chemotherapy in breast cancer and possible targets in drug delivery systems. Pharmaceutics 2020, 12, 1193.
- Izci, M.; Maksoudian, C.; Manshian, B.B.; Soenen, S.J. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem. Rev. 2021, 121, 1746–1803.
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193.
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33.
- Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309.
- Da Silva-Candal, A.; Brown, T.; Krishnan, V.; Lopez-Loureiro, I.; Ávila-Gómez, P.; Pusuluri, A.; Pérez-Díaz, A.; Correa-Paz, C.; Hervella, P.; Castillo, J.; et al. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 2019, 309, 94–105.
- Uhl, C.G.; Gao, Y.; Zhou, S.; Liu, Y. The shape effect on polymer nanoparticle transport in a blood vessel. RSC Adv. 2018, 8, 8089–8100.
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558.
- Rosenholm, J.M.; Sahlgren, C.; Linden, M. Multifunctional mesoporous silica nanoparticles for combined therapeutic, diagnostic and targeted action in cancer treatment. Curr. Drug Targets 2011, 12, 1166–1186.
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337.
- Weaver, J.L.; Tobin, G.A.; Ingle, T.; Bancos, S.; Stevens, D.; Rouse, R.; Howard, D.; Goodwin, K.E.; Knapton, A.; Li, X.; et al. Evaluating the potential of gold, silver, and silica nanoparticles to saturate mononuclear phagocytic system tissues under repeat dosing conditions. Part. Fibre Toxicol. 2017, 14, 25.
- Zhang, Z.; Liu, C.; Bai, J.; Wu, C.; Xiao, Y.; Li, Y.; Zheng, J.; Yang, R.; Tan, W. Silver nanoparticle gated, mesoporous silica coated gold nanorods (@AgNPs): Low premature release and multifunctional cancer theranostic platform. ACS Appl. Mater. Interfaces 2015, 7, 6211–6219.
- Kumar, R.; Mondal, K.; Panda, P.K.; Kaushik, A.; Abolhassani, R.; Ahuja, R.; Rubahne, H.G.; Mishra, Y.K. Core-shell nanostructures: Perspectives towards drug delivery applications. J. Mater. Chem. B 2020, 8, 8992–9027.
- Huo, Q.; Margolese, D.I.; Stucky, G.D. Stucky Surfactant control of phases in the synthesis of mesoporous silica-based materials. Chem. Mater. 1996, 8, 1147–1160.
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S.Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853.
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118.
- Kwon, S.; Singh, R.K.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 2041731413503357.
- Costa, J.A.S.; de Jesus, R.A.; Santos, D.O.; Mano, J.F.; Romao, L.P.C.; Paranhos, C.M. Recent progresses in the adsorption of organic, inorganic, and gas compounds by MCM-41-based mesoporous materials. Microporous Mesoporous Mater. 2020, 291, 109698.
- Oye, G.; Sjöblom, J.; Stöcker, M. Synthesis, characterization and potential applications of new materials in the mesoporous range. Adv. Colloid Interface Sci. 2001, 89–90, 439–466.
- Naono, H.; Hakuman, M.; Tsunehisa, T.; Tamura, N.; Nakai, K. Formation process of MCM-41 precursor and porous texture of MCM-41. J. Colloid Interface Sci. 2000, 224, 358–365.
- Huang, X.; Townley, H.E. An Assessment of mesoporous silica nanoparticle architectures as antigen carriers. Pharmaceutics 2020, 12, 294.
- Wang, S.; Wu, D.; Sun, Y.; Zhong, B. The synthesis of MCM-48 with high yields. Mater. Res. Bullet. 2001, 36, 1717–1720.
- Nandiyanto, A.B.D.; Kim, S.G.; Iskandar, F.; Okuyama, K. Synthesis of spherical mesoporous silica nanoparticles with nanometer-size controllable pores and outer diameters. Microporous Mesoporous Mater. 2009, 120, 447–453.
- Heikkilä, T.; Salonen, J.; Tuura, J.; Hamdy, M.S.; Mul, G.; Kumar, N.; Salmi, T.; Murzin, D.Y.; Laitinen, L.; Kaukonen, A.M.; et al. Mesoporous silica material TUD-1 as a drug delivery system. Int. J. Pharm. 2007, 331, 133–138.
- Kalbasi, J.R.; Zirakbash, A. Synthesis, characterization and drug release studies of poly(2-hydroxyethyl methacrylate)/KIT-5 nanocomposite as an innovative organic–inorganic hybrid carrier system. RSC Adv. 2015, 5, 12463–12471.
- Chircov, C.; Spoială, A.; Păun, C.; Cracium, L.; Ficai, D.; Ficai, A.; Andronescu, E.; Turcule, S.C. Mesoporous silica platforms with potential applications in release and adsorption of active agents. Molecules 2020, 25, 3814.
- Cai, Q.; Luo, Z.S.; Pang, W.Q.; Fan, Y.W.; Chen, X.H.; Cui, F.Z. Dilute solution routes to various controllable morphologies of MCM-41 silica with a basic medium. Chem. Mater. 2001, 13, 258–263.
- Fowler, C.E.; Khushalani, D.; Lebeau, B.; Mann, S. Nanoscale materials with mesostructured interiors. Adv. Mater. 2001, 13, 649–652.
- Nooney, R.I.; Thirunavukkarasu, D.; Chen, Y.M.; Josephs, R.; Ostafin, A.E. Synthesis of nanoscale mesoporous silica spheres with controlled particle size. Chem. Mater. 2002, 14, 4721–4728.
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459.
- Lin, Y.S.; Haynes, C.L. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J. Am. Chem. Soc. 2010, 132, 4834–4842.
- Williams, S.; Neumann, A.; Bremer, I.; Su, Y.; Dräger, G.; Kasper, C.; Behrens, P. Nanoporous silica nanoparticles as biomaterials: Evaluation of different strategies for the functionalization with polysialic acid by step-by-step cytocompatibility testing. J. Mater. Sci. Mater. Med. 2015, 26, 125.
- Qiao, Z.A.; Zhang, L.; Guo, M.; Liu, Y.; Huo, Q. Synthesis of mesoporous silica nanoparticles via controlled hydrolysis and condensation of silicon alkoxide. Chem. Mater. 2009, 21, 3823–3829.
- Blin, J.L.; Michaux, F.; Stébé, M.J. Nanostuctured mesoporous materials from different silica sources using fluorinated surfactants as templates. Colloids Surf. A Physicochem. Eng. Asp. 2016, 510, 104–112.
- Brevet, D.; Jouannin, C.; Tourné-Péteilh, C.; Devoisselle, J.M.; Vioux, A.; Viau, L. Self-encapsulation of a drug-containing ionic liquid into mesoporous silica monoliths or nanoparticles by a sol-gel process. RSC Adv. 2016, 6, 82916–82923.
- Siddiqui, B.; Rehman, A.; Haq, I.; Al-Dossary, A.A.; Elaissari, A.; Ahmed, N. Exploiting recent trends for the synthesis and surface functionalization of mesoporous silica nanoparticles towards biomedical applications. Int. J. Pharm. X 2022, 4, 10016.
- Wang, Y. Synthesis and formation of hierarchical mesoporous silica network in acidic aqueous solutions of sodium silicate and cationic surfactant. Colloid J. 2010, 72, 737–742.
- Das, D.; Yang, Y.; O’Brien, J.S.; Breznan, D.; Nimesh, S.; Bernatchez, S.; Hill, M.; Sayari, A.; Vincent, R.; Kumarathasan, P. Synthesis and physicochemical characterization of mesoporous SiO2 nanoparticles. J. Nanomater. 2014, 2014, 62.
- Li, Y.; Bastakoti, B.P.; Imura, M.; Tang, J.; Aldalbahi, A.; Torad, N.L.; Yamauchi, Y. Dual soft-template system based on colloidal chemistry for the synthesis of hollow mesoporous silica nanoparticles. Chem. Eur. J. 2015, 21, 6375–6380.
- Gai, F.; Zhou, T.; Chu, G.; Li, Y.; Liu, Y.; Huo, Q.; Akhtar, F. Mixed anionic surfactant-templated mesoporous silica nanoparticles for fluorescence detection of Fe3+. Dalton Trans. 2016, 45, 508–514.
- She, X.; Chen, L.; Velleman, L.; Li, C.; Zhu, H.; He, C.; Wang, T.; Shigdar, S.; Duan, W.; Kong, L. Fabrication of high specificity hollow mesoporous silica nanoparticles assisted by Eudragit for targeted drug delivery. J. Colloid Interface Sci. 2015, 445, 151–160.
- Um, K.; Chang, H.; Lee, K. Facile synthesis of hollow mesoporous zinc silicate nanoparticles using a dual surfactant system. RSC Adv. 2016, 6, 98717–98721.
- Napierska, D.; Thomassen, L.C.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39.
- Martin, K.R. The chemistry of silica and its potential health benefits. J. Nutr. Health Aging 2007, 11, 94–97.
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 2017, 29, 1604634.
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108.
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C. Donald Maxwell Parkin Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917.
- Malik, P.; Mukherjee, T.K. Recent Progress in gold and silver nanoparticles biomedical attributes towards lung and breast cancer treatment. Int. J. Pharm. 2018, 553, 483–509.
- Rizwanullah, M.; Ahmad, M.Z.; Ghoneim, M.M.; Alshehri, S.; Imam, S.S.; Md, S.; Alhakamy, N.A.; Jain, K.; Ahmad, J. Receptor-mediated targeted delivery of surface-modified nanomedicine in breast cancer: Recent update and challenges. Pharmaceutics 2021, 13, 2039.
- Duo, Y.; Li, Y.; Chen, C.; Liu, B.; Wang, X.; Zeng, X.; Chen, H. DOX-loaded pH-sensitive mesoporous silica nanoparticles coated with PDA and PEG induce pro-death autophagy in breast cancer. RSC Adv. 2017, 7, 39641–39650.
- Kumar, P.; Tambe, P.; Paknikar, K.M.; Gajbhiye, V. Folate/N-acetyl glucosamine conjugated mesoporous silica nanoparticles for targeting breast cancer cells: A comparative study. Colloids Surf. B 2017, 156, 203–212.
- Ren, W.; Iqbal, M.Z.; Zeng, L.; Chen, T.; Pan, Y.; Zhao, J.; Yin, H.; Zhang, L.; Zhang, J.; Li, A.; et al. Black TiO2 based core–shell nanocomposites as doxorubicin carriers for thermal imaging guided synergistic therapy of breast cancer. Nanoscale 2017, 9, 11195–11204.
- Shen, Y.; Li, M.; Liu, T.; Liu, J.; Xie, Y.; Zhang, J.; Xu, S.; Liu, H. A dual-functional HER2 aptamer-conjugated, pH-activated mesoporous silica nanocarrier-based drug delivery system provides in vitro synergistic cytotoxicity in HER2-positive breast cancer cells. Int. J. Nanomed. 2019, 14, 4029–4044.
- Ali, O.M.; Bekhit, A.A.; Khattab, S.N.; Helmy, M.W.; Abdel-Ghany, Y.S.; Teleb, M.; Elzoghby, A.O. Synthesis of lactoferrin mesoporous silica nanoparticles for pemetrexed/ellagic acid synergistic breast cancer therapy. Colloids Surf. B 2020, 188, 110824.
- Xu, P.; Yao, J.; Li, Z.; Wang, M.; Zhou, L.; Zhong, G.; Zheng, Y.; Li, N.; Zhai, Z.; Yang, S.; et al. Therapeutic effect of doxorubicin-chlorin E6-loaded mesoporous silica nanoparticles combined with ultrasound on triple-negative breast cancer. Int. J. Nanomed. 2020, 15, 2659–2668.
- Moodley, T.; Singh, M. Polymeric mesoporous silica nanoparticles for combination drug delivery in vitro. Biointerface Res. Appl. Chem. 2021, 11, 11905–11919.

