Breast cancer (BC) currently occupies the second rank in cancer-related global female deaths. Although consistent awareness and improved diagnosis have reduced mortality, late diagnosis and resistant response still limit the therapeutic efficacy of chemotherapeutic drugs (CDs), leading to relapse with consequent invasion and metastasis. Treatment with CDs is indeed well-versed but it is badly curtailed with accompanying side effects and inadequacies of site-specific drug delivery. As a result, drug carriers ensuring stealth delivery and sustained drug release with improved pharmacokinetics and biodistribution are urgently needed. Core–shell mesoporous silica nanoparticles (MSNPs) have been a cornerstone in this context, attributed to their high surface area, low density, robust functionalization, high drug loading capacity, size–shape-controlled functioning, and homogeneous shell architecture, enabling stealth drug delivery.
- breast cancer
- chemotherapeutic drugs
- metastasis
- mesoporous silica nanoparticles
- drug delivery
- pharmacokinetics
- sustained release
- biodistribution
1. Introduction
2. Exclusive Terminologies and Synthesis Methods of Mesoporous Silica Nanoparticles
Structurally and morphologically, the MSNPs are solid entities with pores enriched on their surface. “Mesoporous” in MSNPs was a term used to describe pore size. As per the International Union of Pure and Applied Chemistry (IUPAC) regulatory guidelines, porous materials may be microporous, mesoporous, or macroporous. Although their synthesis dates back to the 1970s, Mobil Research and Development Corporation created the first mesoporous solids from aluminosilicate gels in 1992. The associated industrialists labeled it as “Mobil Composition of Matter” or “Mobil Crystalline Materials” (MCM) [24,25][21][22]. Relative distinctions amongst micro, meso, and macroporous are primed exclusively vis-à-vis pore diameters, with <2 nm, (2–50) nm and >50 nm domains. The prominent drug carrier-suited aspects of MSNPs comprise their alterable particle and pore sizes, high stability, rigid structure, large SA, high PV, and robust functionalization and fabrication methods [26,27][23][24]. MSNPs belong to the Exxon Mobil-based M41S group functional configurations, prepared under alkaline conditions in the presence of alkylammonium surfactants and a 3–10 nm pore-sized silica source [28][25]. The functional physicochemical aspects of MSNPs have been demonstrated as significantly varied, via specific choice of initiating precursors, template agents, and reactions. Characterizing aspects of these are their pore sizes (permeability regulators), overall morphology, pore volume and specific SA. Studies demonstrate MCM-41, MCM-48 and MCM-50 as the prominent variants of the M41S family, characterized, respectively, by a hexagonal arrangement, cubic configuration and lamellar morphology [26,29][23][26]. MCM-41 is fabricated using cationic surfactants within a 8.5–12 pH range [30][27]. The most common pore architecture is hexagonal, wherein the cationic-surfactant-to-silica-source (CSS) proportion remains <1 [31][28]. Similarly, MCM-48 and MCM-50 were made by varying the CSS stoichiometry. A cubic pore structure of MCM-48 was retrieved when the CSS ratio was >1 [32][29]. When the stoichiometry was increased further, MCM-50 gradually provided pore structures that resembled lamellar sheets [29][26]. Other mesostructured materials such as Santa Barbara Amorphous (SBA) types were synthesized at the University of California and Technische Universiteit Delft (TUD-1) was developed at Delft University of Technology; COK-12 was synthesized at the Center for Research Chemistry and Catalysis; HMM-33 (Hiroshima Mesoporous Material-33, Japan); and KIT-5 was synthesized at the Korean Advanced Institute of Science and Technology having varied pore sizes and symmetry [33,34,35][30][31][32]. Compared to other members of the MCM family, SBA exhibits a hexagonal 2D structure with thicker pore walls and larger pore size. The SBA mesostructured materials, such as SBA-11, SBA-12, SBA-15 and SBA-16, were created using non-ionic triblock copolymers as templates, such as poly(alkylene oxide) block copolymers and alkyl poly(ethylene oxide) (PEO) oligomeric surfactants [36][33]. These materials prevail in cubic, 3D hexagonal, 2D hexagonal and cubic cage pore symmetry.2.1. Synthesis Methods
Preparation of MSNPs is characterized by the requirements of (i) the silica precursor, the SiO2 source, which forms the walls around the pores, (ii) a surfactant, which aids in the formation of pores, and (iii) an acid or base catalyst, which controls the morphology of the silica structures. The inception of MSNPs’ formation emerges from the sustained interactions of surfactant and silica sources, which provides mesoporous textures. The first successful MSNP synthesis was reported by the Cai, Mann and Ostafin groups, wherein the MSN acronym was coined by Victor Lin to signify the mesoporous silica nanospheres [37,38,39,40][34][35][36][37]. Common SiO2 sources used herein include tetraethyl orthosilicate (TEOS), tetramethyl orthosilicate (TMOS), tetra methoxy vinylsilane (TMVS), tetra butoxy silane (TBOS), tetrakis (2-hydroxyethyl) orthosilicate (THEOS), tetra propyl ortho-silicate (TPOS), trimethoxy silane (TMS) and sodium metasilicate (Na2SiO3). Noted catalysts in the process include NaOH, HCl, NH3, diethanolamine (DEA) and triethanolamine (TEA) [27,41,42,43,44,45,46,47,48][24][38][39][40][41][42][43][44][45]. The surfactant used in the MSNPs’ synthesis is the reason for the varied morphologies, from dispersed monospheres to agglomerates, manifested from altered TEOS hydrolysis and CTAB micellization. The prevalence of agglomerates in the solution phase relates to a threshold surfactant extent, which varies with the mutual hydrophobicity of the silica precursor. Familiar surfactants used as templates in MSNPs are cetyltrimethylammonium chloride (CTAC), cetyl trimethyl ammonium bromide (CTAB), hexadecyltrimethylammonium (HDTMA), sodium dodecyl benzene sulfonate (SDBS), sodium dodecyl sulfate (SDS), Triton X-100, polysorbate, Pluronic F127, Pluronic P123, and phospholipids, betaines and amino acids [49,50,51,52][46][47][48][49]. The key parameters affecting the formation of MSNPs are the reaction mixture’s pH, the nature of the surfactants/copolymers, in addition to the relative extents and sources of silica. Prominent synthesis methods are hereby distinguished as sol–gel and hydrothermal and a contemporary green method, the working aspects of which are discussed ahead (Figure 3).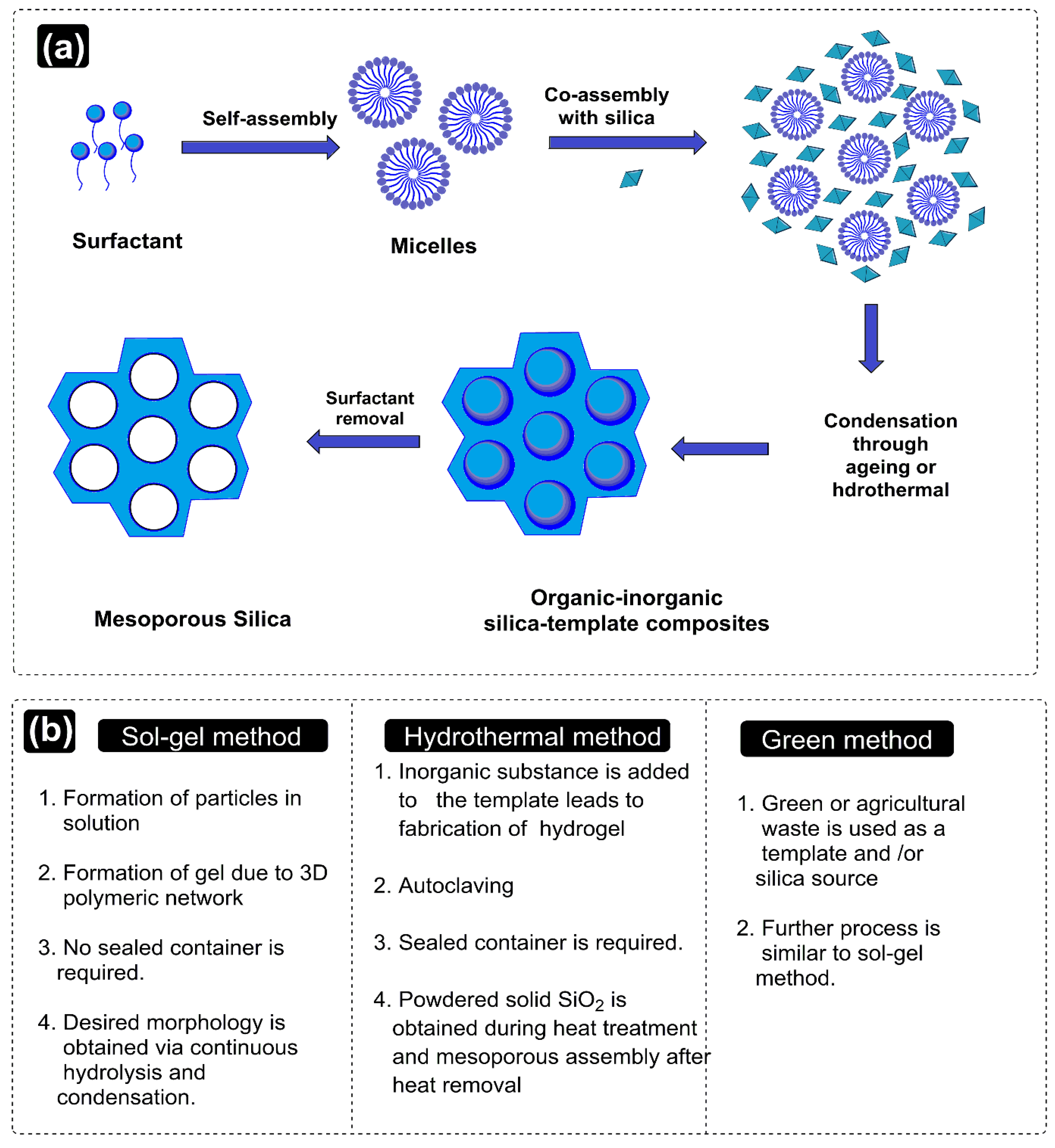
3. Promising Aspects of Mesoporous Silica Nanoparticles as Drug Vehicles
4. Drug Release Mechanisms of Mesoporous Silica Nanoparticles
As is the case with most drug-delivering NPs, MSNPs could also be tuned for active or passive drug delivery. The former of these mechanisms relies on the proximity to surface ligands, while the passive regime exploits the troubled physicochemical vicinity of tumor cells for tumor cell drug internalization. Figure 4 depicts the functional distinctions of ligands frequently used for MSNP-aided drug delivery to tumor cells. It is worth noting here that a delivery regimen could be programmed via dual ligand binding, depending on the tumor severity. Contrary to the above, the passive mode relies on enhanced permeation and retention (EPR), gathering the threshold drug extent in the tumor cell vicinity. Although the active mode is more accurate, its optimization is comparatively stringent (ligand topography and distribution), alongwith a greater risk vis-à-vis localized pH or temperature changes. Several comprehensive reviews discuss the mechanistic distinctions of active and passive delivery regimes, pictorially distinguished in Figure 5. Unlike Au and Ag NPs, MSNPs exhibit significant functionalization diversity, whereby the role of surface pores, gatekeeping-driven drug release and PVs together assume a significant contribution.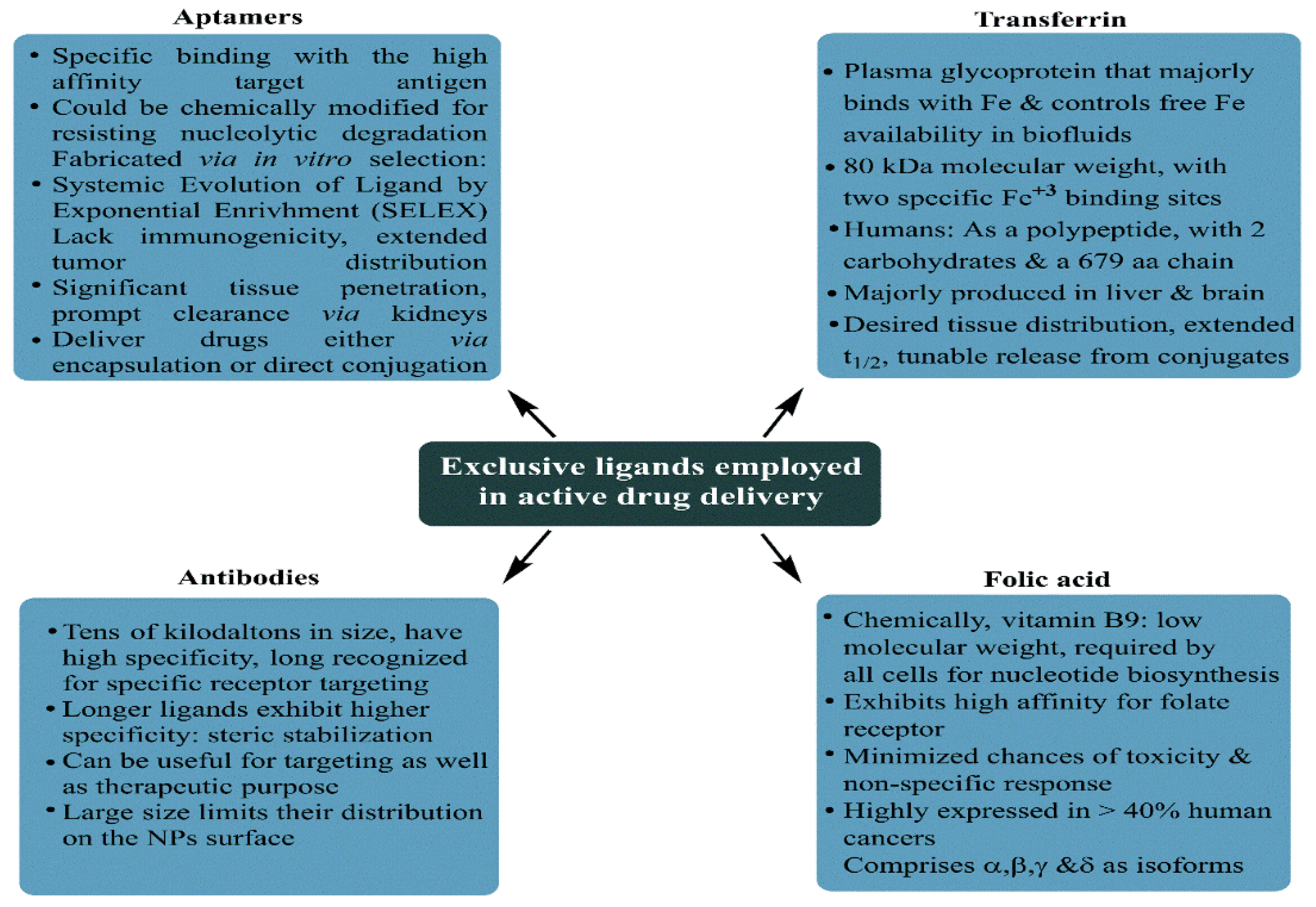
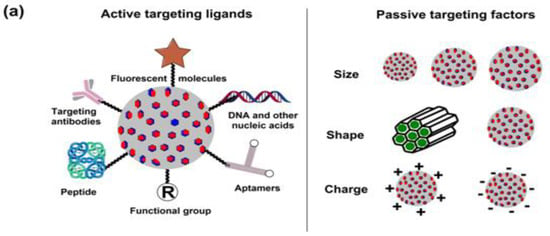
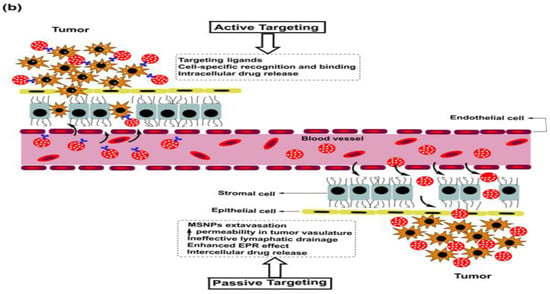
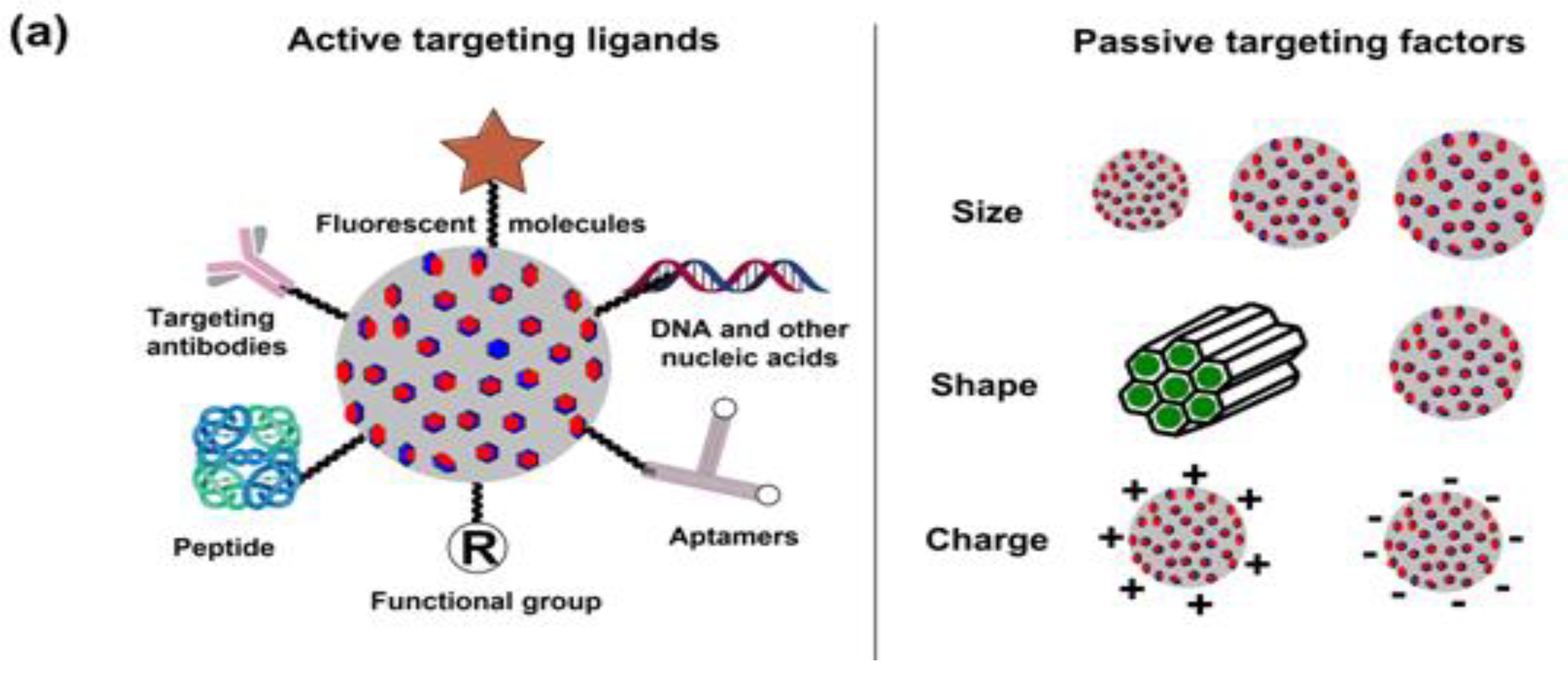
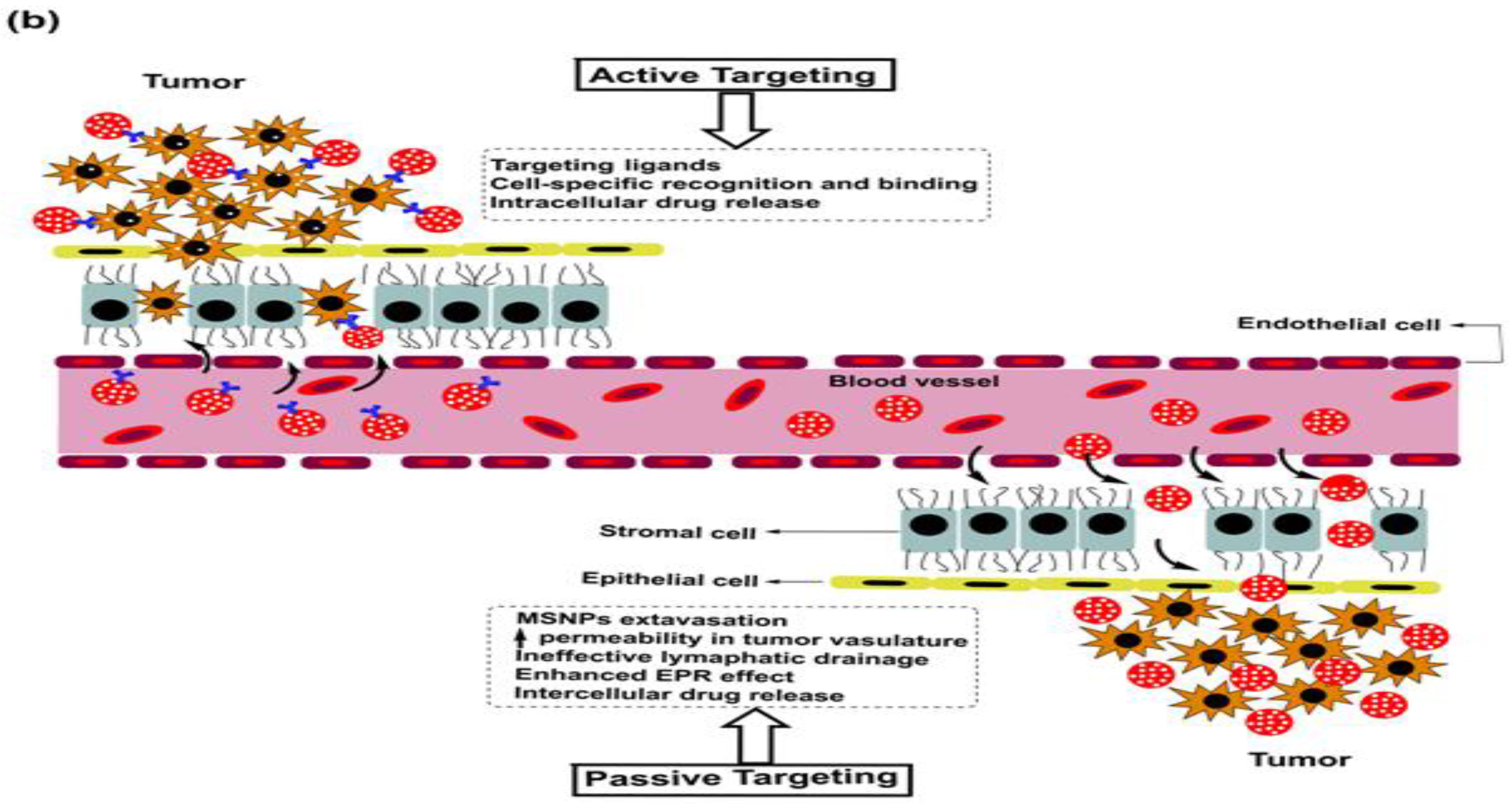
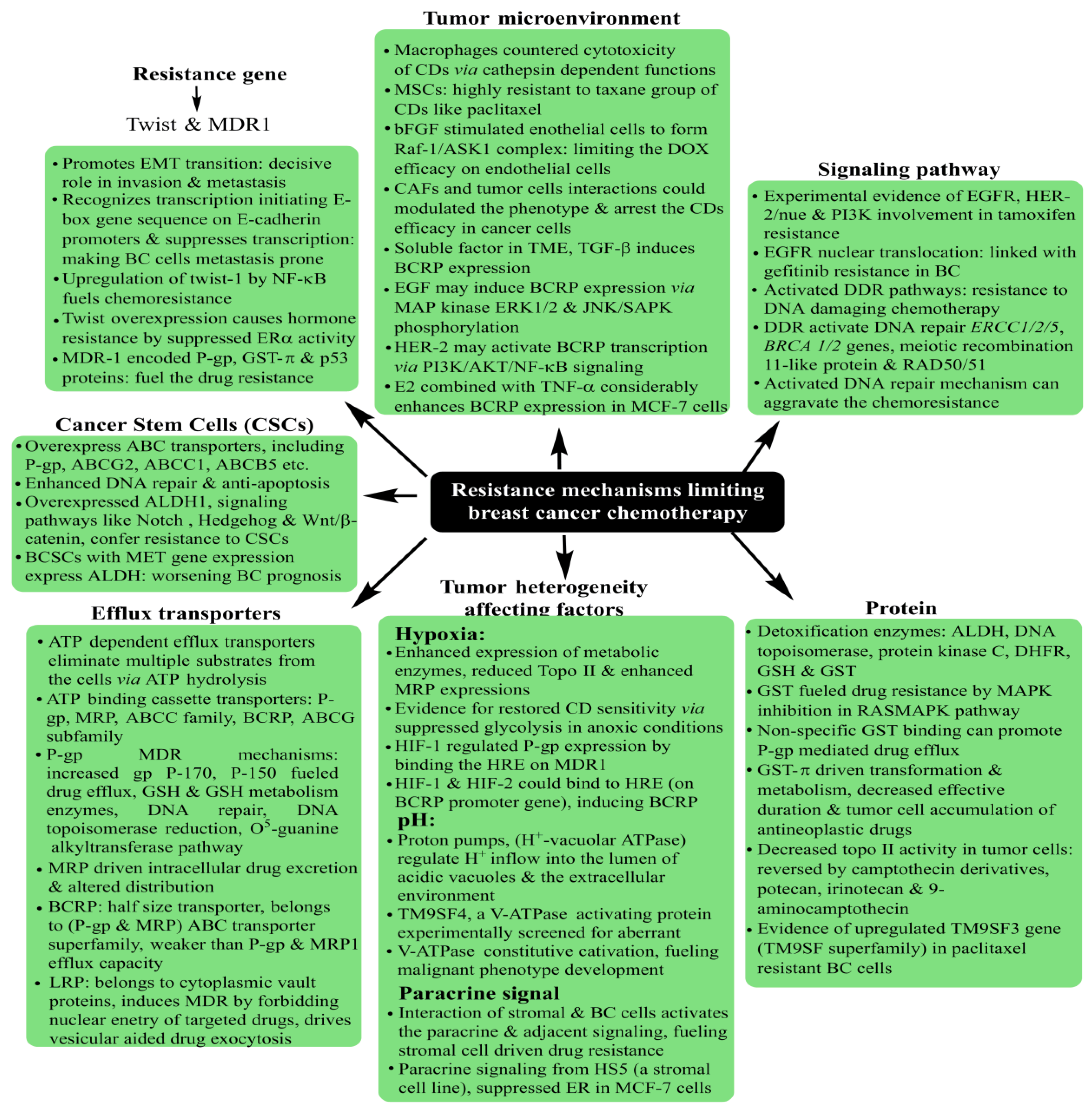
5. Breast Cancer: Pathophysiology, Mortality and Chemoresistance
Prevailing as the most threatening cancer affecting females, BC has been a major health concern for more than a decade now, with 2012 GLOBOCAN statistics registering a staggering 18% rise in casualties compared to the number of 2008 cases [165,166][53][54]. A predictive analysis by the American Cancer Society anticipates one out of every eight United States females to develop BC in her lifetime. On a global platform, too, trends reflect a significant concern, with an addition of 3.2 million new cases per year until 2050. As rising mortality concerns continue unabated, it is a priority to screen accurate and reliable biomarkers, in addition to timely diagnosis, to improve the success of preventive treatment (Figure 6). Development in diagnostic assays in the past few years has simplified the treatment rigor, whereby rising mortality has witnessed a substantial arrest. Global awareness campaigns and recent research findings have enhanced the unanimity for BC classification, vis-à-vis characteristic histology and molecular events.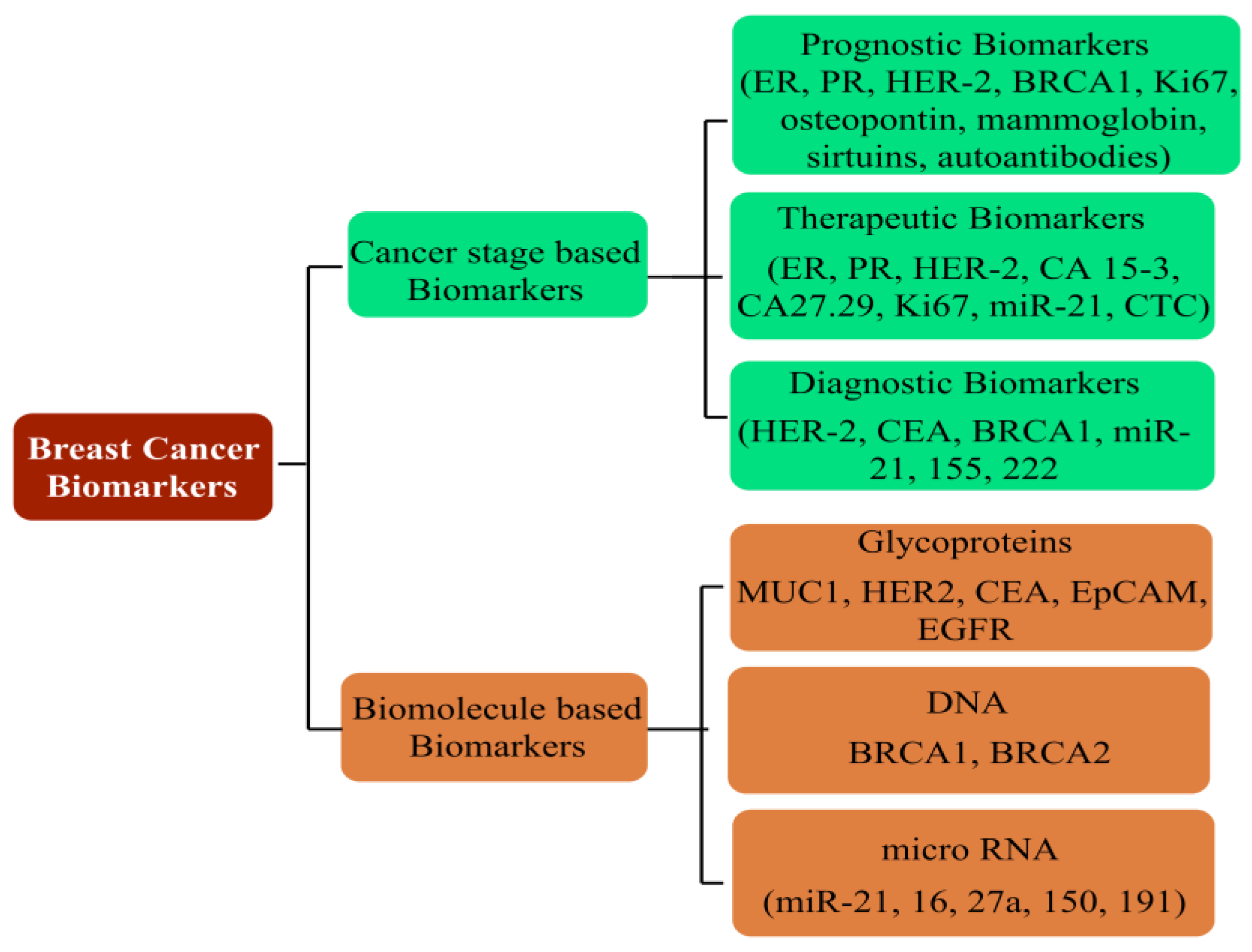
Name of Receptor | Physiological Distinctions | Potential in Breast Cancer Treatment | ||||||
|---|---|---|---|---|---|---|---|---|
ErbB tyrosine kinase receptors | Widely investigated growth factor receptors in BC that consist of four homolog receptors: ErbB1 (HER1/EGFR), ErbB2 (HER2/neu), ErbB3 (HER3) and ErbB4 (HER4). EGFR and HER2 are overexpressed in 15–20% and 20–25% of BC cells. The BC cells overexpressing ErbB receptors demonstrate aggressive clinical behavior. | Enhanced cytotoxicity of CURC delivered via EGFR targeting GE11 peptide-conjugated CURC-loaded PLGA-PEG NPs (in MCF-7 cells). HER2 Ab-conjugated PTX and rapamycin-loaded glycerol monooleate-coated magnetic NPs exhibited enhanced uptake in MCF-BC cells with 24-fold reduced toxicity and 3-fold lower IC50 extent (for PTX). For rapamycin, 71-fold reduced IC50 than anative drug with targeted magnetic NPs. EGFR-conjugated immuno NPs exhibited ~13-fold higher uptake and antiproliferative activity than unconjugated NPs in MCF-7 BC cells. HER2-targeted PLGA/montmorillonite-trastuzumab NPs exhibited 12.74 and 13.11-fold larger therapeutic efficacy than untargeted NPs. | ||||||
Folate receptor | Binds to water-soluble, low-molecular-weight FA (vitamin B9), essential in normal mitotic cell division;, binds to the folate ligand and then internalizes the complexes via receptor-mediated endocytosis. | Studied for high anticancer efficacy of PTX (in 4T1 BC cells), cisplatin (DDP) and docetaxel (in MDA-MB-231 BC cells), CURC (in MCF-7 cells), enhanced cellular uptake for deoxycholic acid-O-carboxymethylated CTS and vincristine sulfate-loaded PLGA-PEG NPs | ||||||
Estrogen receptors | Belong to thenuclear hormone receptor superfamily, differentially overexpressed in 60–80% of BC cells, internalized in the cancer cells upon binding to ERs | Enhanced cellular uptake of PEG-conjugated PTX-epirubicin co-loaded liposomal NPs, TAM surface-grafted liposomes loaded with DOX (in MCF-7 cells), DOX and DOX + TAM-loaded liposomes (MCF-7 cells) | ||||||
CD44/Hyaluronan receptor | A natural component of ECM, hyaluronan is critically involved in cell proliferation, migration and invasion. CD44 regulates lymphocyte adhesion to endothelial cells during lymphocyte migration (a process equivalent to metastasis in solid tumors). CD44 prevails as an HA receptorandprevails sparsely on the surface of epithelial, hematopoietic and neuronal cells. Recent studies have screened CD44 as a BC stem cell marker. | HA-functionalized CTS lipoic acid NPs loaded with 17α-methyltestosterone exhibited enhanced internalization in CD44 overexpressing BT-20 BC cells than CD44-negative MCF-7 cells. Enhanced uptake and cytotoxicity of PLGA NPs in MDA-MB-231 cells. Higher docetaxel uptake for self-assembled PLGA-HA block copolymers (in MDA-MB-231 cells) than untargeted NPs. HA matrix NPs with intrinsic CD-44 tropism and loaded rapamycin exhibited a 3.2-fold enhanced uptake in CD44-positive MDA-MB-468 cells. HA-lysine-lipoic acid NPs loaded with DOX exhibited a 20-fold enhanced uptake in MCF-7/ADR cells. | ||||||
Luteinizing hormone-releasing hormone receptor (LHRH) | A hormonal decapeptide produced by the hypothalamus, LHRH is also known as a gonadotropin-releasing hormone. Plays a key role in regulating the pituitary–gonadal axis and reproduction. Overexpressed in 50% of BC cells. | LHRH-conjugated PTX showed enhanced specificity for targeting MDA-MB-231 cells. LHRH-CTS-conjugated NPs enhanced cellular uptake with two-fold reduced IC50in LHR overexpressing MCF-7 cells than non-targeted NPs. DDP-loaded LHRH-modified dextran NPs exhibited a higher uptake with reduced nephrotoxicity of DDP than non-targeted NPs (in 4T1 BC cells). |
6. Discussion of Recent Attempts Using Mesoporous Silica Nanoparticles for Breast Cancer Treatment
This section discusses the major findings of recent studies (2017 onwards) using MSNPs for BC treatment, distinguished via singular and dual therapeutic agents. Emphasis has been laid on comparative IC50 reductions, moderated drug release kinetics and enhanced tumor cell internalization abilities. The advantage of dual therapeutic agents is exhibited by the stoichiometric variation of a combinatorial mode, involving the structural changes that decrease the chances of obtaining a resistant response. Although MSNPs do prevail as a carrier in both modes, the therapeutic efficacy of a combined mode is generally higher (likely due to a synergistic influence), owing to which substantial IC50 reductions could be accomplished.6.1. Singular Drug Delivery Using Mesoporous Silica Nanoparticles
Commencing from 2017, the first major effort used polydopamine (PDA) and polyethylene glycol (PEG)-coated MSNPs for DOX delivery to MCF-7 and MDA-MB-231 BC cells. The PDA coating enabled a pH-mediated DOX release, while the PEG functionalization conferred an improved physiological biocompatibility. Dispersion profile analysis for the MSNPs in neat, DOX-loaded, PDA and PEG coated states, revealed increments and decrements in PS and PVs. Though PDA and PEG-coated MSNPs exhibited pore sizes of ~200 nm, which were smaller than the cut-off limits of tumor neovasculature pores and exhibited suitability for the EPR effect. Anionic surface sensitivity guarded against reticuloendothelial clearance, facilitating an elongated physiological residence. The inspection of the morphology using Transmission Electron Microscopy (TEM) implied a spherical morphology with porous surfaces for all states of the MSNPs, conveying the structural compatibility of PDA and PEG coating. These physicochemical features together contributed to a ~95% DOX encapsulation efficacy (EE). An analysis via confocal microscopy and flow cytometry for cellular uptake distinctions revealed enhanced extents for PDA and PEG-coated, DOX-loaded MSNPs compared to free DOX. These observations were supported by the MTT assay, which implied higher cell growth and proliferation inhibition for PDA and PEG-coated, DOX-loaded MSNPs than free DOX and the NPs without PEG coating. The antitumor mechanism of intact MSNPs was screened using fluorescence analysis, wherein MSNP–DOX–PDA–PEG-treated MCF-7 cells formed greater autophagic vesicles (green fluorescence) over other configurations. The regulatory influence of the autophagic response was screened via assessing the AKT, mTOR and p70S6K phosphorylations, with MSNP–DOX–PDA–PEG causing the maximum inhibition. The observations were replicated in the nude mice harboring subcutaneous MCF-7 tumors, with MSNPs–DOX–PDA–PEG exhibiting the maximum inhibition in terms of maximum tumor weight decrements. Furthermore, no significant distinctions in the body weight and histopathological abnormalities (heart, liver, spleen, kidney and lungs) for all the carrier configurations imply the biocompatibility of MSNP–DOX–PDA–PEG [169][57]. This restudyearch, therefore, optimized the pH-specific DOX release kinetics via PDA and PEG–MSNP functionalization, conferring high tolerability with enhanced tumor cell uptake. A subsequent effort in 2017 used folic acid (FA) and N-acetyl glucosamine (NAG)-functionalized MSNPs for DOX delivery to MCF-7 and MDA-MB-231 human BC cells. With a 78 nm PS and a cationic surface sensitivity (unlike the previous study), the prepared MSNPs exhibited compatibility for EPR-driven tumor cell internalization aided by electrostatic receptivity with negatively charged membrane lipids. Monodispersed states with the spherical morphology of prepared MSNPs were inferred by Scanning Electron Microscopy (SEM) analysis, while TEM screening implied their sieve-resembling structure, harboring nanoporous channels on the surface. No structural abnormality was observed for FA and NAG functionalizations in the FTIR screening, but a thermal stability inspection using thermogravimetric inspection revealed a 10% and 19% higher weight loss for NAG and FA-functionalized MSNPs compared to their neat state. This weight loss was attributed to the moderated NAG and FA contributions, with nearly twice the FA molecular weight contributing to its higher loss. Cellular uptake studies using confocal microscopy and fluorescence spectroscopy (comparative red and green intensities) both implied the significant uptake of NAG and FA-functionalized, DOX-loaded nanocarriers (NCs). Quantitative screening by flow cytometry revealed 52.36 ± 3.98 and 60.1 ± 9.37% internalizations for FA and NAG-functionalized NPs in MCF-7 cells, while similar amounts for MDA-MB-231 cells were 52.74 ± 1.08 and 60.58 ± 6.03%. The ligand conjugation of DOX-loaded NCs enhanced their toxicity, as screened by the MTT assay, with 48 and 72 h viability for 100 μg·mL−1 extents being 10.68 ± 0.59 and 4.34 ± 0.45% for DOX–NAG–MSNPs and 14.57 ± 0.65 and 8.95 ± 1.61% for DOX–FA–MSNPs. Similar extents for more aggressive MDA-MB-231 cells were 5.41 ± 0.12 and 5.15 ± 0.41% for FA-functionalized and 7.90 ± 1.67 and 5.76 ± 0.31% for NAG-functionalized DOX-loaded NPs. The NAG functionalization exhibited greater cytotoxicity (IC50 = 0.86 μM) than FA (IC50 = 2.5 μM), which was explained by investigators to be due to a comparatively higher glucose transporter (GLUT) expression on BC cells’ surface compared to the folate receptor α (FRα). The mechanism of the anticancer response was screened using AO/EB fluorescent staining, with minimal necrotic cells exhibiting varied orange-red EB fluorescence, while AO penetrated the intact membrane cells and developed green fluorescence. These distinctions implied apoptosis to be the likely source of cell death following exposure to drug-loaded MSNPs [170][58].6.2. Combinatorial Drug Delivery Using Mesoporous Silica Nanoparticles
Recurrent resistance with CDs has reduced their clinical efficacy, thereby mandating a need for shifting to potential alternatives via predictive structure–activity relationships (SARs). In this regard, the combinatorial delivery of CDs (two or more) with sensitization agents (such as photothermal or X-ray irradiation) has lately gathered significant interest. The turning point aspect revolves around the long-known and practiced status of most of the CDs due to which their continued use inevitably receives a resistant response despite a stealth delivery via various NCs. To counter the competing therapeutic limitations, combining two or more CDs or the sensitizing agents with CDs has lately been an area of focus for developing potent drug delivery vehicles. This strategy benefits from multiple considerations, wherein the most important distinction is the reduced elimination on an in vivo scale. Newer combinations are not recognized as native, unlike the long-practiced CDs, whereby there stands a significant possibility of these being more effective. Secondly, for terminally ill or advanced stages (as in TNBC), rapid metastasis and fragility mandate accurate therapies. Using the combinatorial route, the concentration of the major CDs (often resistant) is effectively reduced, wherein the sensitization of healthy cells is substantially reduced. The combination of drugs no longer targets a single receptor on tumor cells and its unique chemical essence is the tunability with the mutated status of receptor proteins. Thereby, the chances of prompt elimination from physiological boundaries are much lower for a combinatorial regime. The most important benefit of combinatorial delivery is the possible use of bioactive and non-toxic nutraceuticals. Therefore, delivering a small proportion of CDs with these compounds could significantly improve their tumor cell internalization. Stronger therapeutic essence and dosage moderation are inevitable via NC-mediated delivery, as additional structural protection is accomplished. As for MSNPs, recent interest has superseded the success of Au and Ag NPs, both of which lack the porous morphology, and Ag NPs also exhibit high native toxicity that complicates their systemic elimination. Keeping these prospects in the background, the recent attempts (since 2017) using MSNPs for combinatorial drug delivery to treat BC are summarized in the following paragraphs. The single significant attempt from 2017 used TiO2-coated MSNPs as DOX carriers, with the assistance of 808 nm near-infrared (NIR) irradiation for thermal imaging-aided photothermal therapy (PTT) for BC chemotherapy. The study by Ren and colleagues highlighted the constraints of traditional white TiO2-like weak drug loading, inadequate UV light penetration in the tissues and the heating risks of 980 nm NIR to the healthy tissues. The preliminary screening of black TiO2 (B-TiO2) NPs revealed a PS of 25 nm (via TEM) with weak dispersion. Coating these NPs with mesoporous silica conferred a core–shell morphology to B-TiO2 NPs with a ~100 nm diameter. The prepared nanocomposites (B-TiO2 NPs in an MSNP core) were functionalized by the -NH2 group and subsequently reacted with-COOH in the FA, using an EDC–NHS conjugation. The recovered NCs were loaded with DOX, exhibiting a 150–260 nm PS and a <1 PDI for B-TiO2, NC, NC–NH2, NC–FA and NC–FA–DOX. The –NH2, FA and DOX conjugation to the NC surface was distinguished via the cationic surface sensitivity for B-TiO2 and NC–NH2comparedto anionic sensitivity for others. The DOX loading on NCs was screened as 5%, ~10-fold higher than uncoated B-TiO2. Screening the photothermal irradiation impact on DOX release from NCs, NIR irradiation at pH 5 enhanced the release from 60.6% (neat) to 91.3% in 48 h, conveying a pH and NIR-modulated liberation. The impact of FA targeting was distinguished in the fluorescence analysis, wherein NC–FA–DOX exhibited DOX deposition in the nucleus, while the non-targeted NCs (without FA) revealed little DOX in the cytoplasm, with inadequate nuclei localization. Screening the PTT influence on the therapeutic efficacy of DOX, a better performance for NC–FA was noticed than for NCs alone, with a 0–5 min NIR exposure on incubation with 75, 150 and 225 μg·mL−1 Ti, generating the 85.2 and 50.8% at 75 μg·mL−1, 76.5 and 10.9% at 150 μg·mL−1 and 34.1 and 5.5% at 225 μg·mL−1 viabilities for NC and NC–FA confirmations. Monitoring the influence of PTT on the anticancer effects of DOX, the analysis revealed cell death of 31.6% (by DOX alone) and merely 8% (by NC–DOX); and with NC–FA–DOX, this extent was 28.7%. On subjecting the treated cells to 5 min NIR irradiation, these limits increased to 44.8, 34.1 and 93.8%, implying the synergism of PTT and chemotherapeutic treatments. Screening the cell death mechanism in NC–FA and NC–FA–DOX-treated cells, a flow cytometry-driven inspection revealed most tumor cells being subjected to necrosis (death via photothermal-induced ablation). The in vivo screening of chemotherapy and PTT synergism was made in tumor-implanted xenograft mice, distinguished as (i) only DOX-treated, and (ii) treated with NC–FA, (iii) NC–FA + NIR irradiation, (iii) NC–FA–DOX and (v) NC–FA–DOX + NIR irradiation. An analysis of five mice, which were sacrificed to consolidate the PTT–chemotherapy synergism using HE staining revealed most cells to be dead in the DOX, NC–FA + NIR, and NC–FA–DOX groups, exhibiting nuclear or cell membrane damage. Greater tumor cell killing in the NC–FA–DOX + NIR group suggested a synergistic combination of PTT and chemotherapy rather than a singular effect. The observations were further strengthened by significant decrements in TV for the NC–FA–DOX + NIR group compared to the NC–FA + NIR and NC–FA–DOX groups. Moreover, no significant variations in body weight revealed the biocompatibility of the used NCs [192][59]. Thereby, this restudyearch established a synergism of TiO2 NPs’ toxicity with the FA-conjugated MSNPs for the localized therapeutic induction in the MCF-7 BC cells. Similar attempts could be made by co-delivering TiO2 NP-doped MSNPs with other CDs and some phytomolecules, which could further moderate the toxicity. Secondly, in place of Ti, the oxides of other elements (in the same group of the periodic table) could be a logical extension of this restudyearch. The year 2019 reported a sole major attempt relying on HER2-conjugated aptamer (HApt) cytotoxicity via cross-linking and subsequent HER2 translocation to cytoplasmic vesicles in the SKBR3 and MCF-7 BC cells. Relying on HER2 suppression-triggered impaired cell proliferation and impaired apoptosis, Shen and colleagues exploited the HApt targeting and antagonizing ability to augment the HER2-positive BC cell-specific toxicity of MSNPs. The carrier configuration involved HApt-functionalized pH-sensitive β-cyclodextrin (β-CD) capped with MSNPs conjugated with benzimidazole (MSN-BM), which was used to optimize a pH-driven DOX release. The purpose of including β-CD was to use its gatekeeper essence to deliver encapsulated DOX-HApt as the HER2-targeting biotherapeutics. The thorough physicochemical characterization of the optimized carrier revealed nanoscale attributes (via FT-IR, XRD, TEM and BET) with no structural arbitration. The DOX release was studied at pH 7.4, 6.4 and 4.5, with the maximum extent (>80%) being accomplished at pH 4.5, due to BM protonation-disrupted DOX–BM hydrophobic interactions. Comparing the NC-delivered DOX efficacy in HER2-positive SKBR3 and HER2-devoid MCF-7 cells, greater cytotoxicity, as well as enhanced uptake, was observed in the SKBR3 cells. The synergistic association was witnessed as the DOX-HApt co-delivery enabled higher toxicity in HER2-positive BC cells compared to either DOX or HApt. The reduction in cell viability for DOX-devoid NCs was15 ± 2%, for 100 μg·mL−1 NPs, while similar results for free DOX and aptamer-devoid NC–DOX (DOX extent: 3.6 μg·mL−1) were 8 ± 4% and 27 ± 6%. The MSN-BM/CD-HApt@DOX, however, reduced the viability by ~68 ± 6%, signifying a DOX and HApt synergistic essence [193][60]. The first major 2020 attempt focused on MSNP-facilitated combinatorial drug delivery to treat BC, using lactoferrin (Lf)-coupled NPs for simultaneous pemetrexed (PMT, a cytotoxic drug) and ellagic acid (a phytocompound) administration to MCF-7 human BC cells. The hydrophobic EA was physically encapsulated within the mesopores (via adsorption) assisted by electrostatic interactions between anionic EA and -NH2 group functionalized NCs. Contrary to this, the hydrophilic PMT was chemically conjugated to Lf coating via carbodiimide coupling, to prevent its early release and instantaneous toxicity aggravation. The dispersion analysis of PMT + EA-loaded NCs attributed their nanoscale distinction, with a PS of 284 nm (via DLS), 190–230 nm (via TEM imaging) anda 0.207–0.778 PDI, enabling a sequential drug release, first of EA, followed by sustained PMT liberation. The co-loading features were demonstrated well via differential scanning calorimetry (DSC) and powder XRD, showing a simultaneous drug loading that resulted in crystallinity loss. Structural coherence was inferred by FTIR spectroscopy with C=O ester of EA (1723 cm−1), C=O functionality of the second –COOH group of PMT (1690 cm−1), and the (1690–1630 cm−1) stretching frequencies of newly formed amide linkages. The synergistic association between PMT and EA was monitored via cytotoxicity studies, wherein 5:3 proportions of unaided EA and PMT exhibited 1.7 and 1.3-fold decrements in IC50 of EA (34 μg·mL−1) and PMT (26 μg·mL−1), complemented by a combination index (CI) of 0.975. These observations were further strengthened for NC-mediated EA delivery with a 23 and 20 μg·mL−1 IC50 for non-targeted and targeted NC configurations. Yet again, a CI value of 0.885 suggested EA and PMT synergism (less so than for unaided delivery). The role of Lf targeting in desired drug internalization in the MCF-7 tumor cells was screened via fluorescence intensity, which developed a faint green appearance, inhibiting the Lf receptors and incubating the cells with excess Lf before the uptake analysis. The results were supported by a flow cytometry inspection, wherein high MFI implied a higher intake of NCs by the tumor cells in the Lf-conjugated state [194][61]. Thereby, this restudyearch established Lf-targeted MSNPs as promising carriers for mediating synergistic PMT and EA anticancer actions in MCF-7 BC cells. The results indeed encourage evaluation in animal models and more aggressive BC subtypes. A subsequent 2020 attempt towards using MSNPs for combinatorial therapies against BC involved DOX and a sonosensitizer, chlorin-e6 (Ce6), thereby combining sonodynamic therapy (SDT) with DOX–MSNPs. The prepared MSNPs (using CTAB as a surfactant and TEOS as a template) were optimized for DOX delivery via initial –NH2 and subsequent –COOH functionalization, which was observed in −31–20, 17–37 and −37 to −43 mV ranges o fζ-potentials for native, –NH2 and –COOH-functionalized states, respectively. The variations suggested progressive -OH and –NH2 replacements, with a higher colloidal stability of –COOH-functionalized MSNPs. The PS of NCs using TEM was screened as 149.5 ± 12.2 nm, with the magnitudes reaching up to 300 nm in DLS measurements, due to conjugated water molecules. The DOX release was monitored without pH fixation, reaching an 80% extent after 10 h, due to the obstruction of the MSNPs’ pores. The release was slow but not sustained due to the NCs’ surface lacking mesopores. Uptake studies deciphered a therapeutic significance of MSN–DOX–Ce6 particles, as these were internalized in MDA-MB-231 cells to a higher extent (greater fluorescence) than DOX and Ce6 alone. The release mechanism was ascertained as “free-diffusion”, with the cells requiring co-culturing with drugs for 4 h before the sonication-energized antitumor efficacy of Ce6. Screening the antitumor efficacy of MSNP–DOX–Ce6, the carriers without DOX caused no change in cell viability, while MSNP–DOX–Ce6 with ultrasound (US), decreased it more than US–Ce6, DOX+Ce6+US or free DOX. The effects were reproduced in vivo (BALB/c nude mice) with substantial TW and TV decrements for the MSNP–DOX–Ce6–US group rather than Ce6–US or DOX alone [195][62]. Thereby, this restudyearch established an MSNP–DOX–Ce6 and US synergism for DOX efficacy in MDA-MB-231 TNBC treatment. In perhaps the last major attempt from 2020, Moodley and Singh used MSNPs neat, in a CTS-functionalized state and with (2–5)% PEG functionalization for DOX and 5-FU co-delivery to MCF-7 BC cells. The as-synthesized MSNPs (CTAB as a template, TEOS as a silica precursor) were functionalized with CTS and PEG. The optimum functionalization was attained by the reversal of the anionic surface sensitivity (for MSNPs) to the cationic regime in the CTS and PEG-functionalized state. The TEM-ascertained PS for MSNPs ranged between 37 and 66 nm, while the 12–215 nm hydrodynamic diameters were due to the enhanced aqueous interactions of the silanol groups. The 710.36 m2·g−1 SA and 1.74 cm3·g−1 PV implied a nanoscale sensitivity of the prepared NPs, prevailing as graduated mesoporous spheres for drug loading and release. Encapsulation of 5-FU and DOX into the PEG-CTS@MSNPs determined a combined drug loading of 20–30%, due to their differential interactions with the carrier constituents. With a higher hydrophobicity, DOX interacted with hydrophilic PEG and was internalized to a greater extent than 5-FU, which is attracted to CTS and –NH2 groups and is internalized afterwards. The release kinetics were monitored at pH 7.4 and 4.2, with the former resulting in a higher 5-FU + DOX liberation of 21% and 52% for NCs functionalized with 2% PEG. At pH 4.2, the release extents were 29 and 59% for 2 and 5% PEG-functionalized NCs and for 5-FU, while for DOX, these were 62 and 53%. The distinctive release extents of 5-FU and DOX were attributed to the pH-dependent 5-FU protonation, wherein a 4.2 pH caused Fickian diffusion for both drugs from 2% PEG-functionalized NCs with negligible erosion. For 5% PEG-functionalized NCs, the diffusion and erosion constants played a complementary role to modulate DOX release. Slower drug diffusion was reasoned by investigators due to DOX interaction with the MSNPs matrix and aqueous medium, besides the pH and PEG coating on the MSNPs. Screening the antitumor efficacy, the MCF-7 BC cells treated with 5-FU + DOX revealed maximum apoptosis with 2% PEG-functionalized NCs, causing DNA condensation and fragmentation. A cell cycle analysis revealed enhanced populations in S and G2/M phases, inducing DNA damage and cell cycle arrest, correlating with most of the cells undergoing apoptosis and fragmentation. These were witnessed in AO/EB-stained cells, hinting at the toxicity emanated by 5-FU and DOX-co-loaded MSNPs [196][63].References
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66.
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.C.; Martin, M.; Namer, M.; et al. The global breast cancer burden: Variations in epidemiology and survival. Clin. Breast Cancer 2005, 6, 391–401.
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434.
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800.
- Muley, H.; Fad, R.; Rodriguez-Rodriguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Phamacol. 2020, 177, 113959.
- Lainetti, P.d.F.; Leis-Filho, A.F.; Laufer-Amorim, R.; Battazza, A.; Fonseca-Alves, C.E. Mechanisms of resistance to chemotherapy in breast cancer and possible targets in drug delivery systems. Pharmaceutics 2020, 12, 1193.
- Izci, M.; Maksoudian, C.; Manshian, B.B.; Soenen, S.J. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem. Rev. 2021, 121, 1746–1803.
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193.
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33.
- Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309.
- Da Silva-Candal, A.; Brown, T.; Krishnan, V.; Lopez-Loureiro, I.; Ávila-Gómez, P.; Pusuluri, A.; Pérez-Díaz, A.; Correa-Paz, C.; Hervella, P.; Castillo, J.; et al. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 2019, 309, 94–105.
- Uhl, C.G.; Gao, Y.; Zhou, S.; Liu, Y. The shape effect on polymer nanoparticle transport in a blood vessel. RSC Adv. 2018, 8, 8089–8100.
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558.
- Rosenholm, J.M.; Sahlgren, C.; Linden, M. Multifunctional mesoporous silica nanoparticles for combined therapeutic, diagnostic and targeted action in cancer treatment. Curr. Drug Targets 2011, 12, 1166–1186.
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337.
- Weaver, J.L.; Tobin, G.A.; Ingle, T.; Bancos, S.; Stevens, D.; Rouse, R.; Howard, D.; Goodwin, K.E.; Knapton, A.; Li, X.; et al. Evaluating the potential of gold, silver, and silica nanoparticles to saturate mononuclear phagocytic system tissues under repeat dosing conditions. Part. Fibre Toxicol. 2017, 14, 25.
- Zhang, Z.; Liu, C.; Bai, J.; Wu, C.; Xiao, Y.; Li, Y.; Zheng, J.; Yang, R.; Tan, W. Silver nanoparticle gated, mesoporous silica coated gold nanorods (@AgNPs): Low premature release and multifunctional cancer theranostic platform. ACS Appl. Mater. Interfaces 2015, 7, 6211–6219.
- Kumar, R.; Mondal, K.; Panda, P.K.; Kaushik, A.; Abolhassani, R.; Ahuja, R.; Rubahne, H.G.; Mishra, Y.K. Core-shell nanostructures: Perspectives towards drug delivery applications. J. Mater. Chem. B 2020, 8, 8992–9027.
- Huo, Q.; Margolese, D.I.; Stucky, G.D. Stucky Surfactant control of phases in the synthesis of mesoporous silica-based materials. Chem. Mater. 1996, 8, 1147–1160.
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S.Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853.
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118.
- Kwon, S.; Singh, R.K.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 2041731413503357.
- Costa, J.A.S.; de Jesus, R.A.; Santos, D.O.; Mano, J.F.; Romao, L.P.C.; Paranhos, C.M. Recent progresses in the adsorption of organic, inorganic, and gas compounds by MCM-41-based mesoporous materials. Microporous Mesoporous Mater. 2020, 291, 109698.
- Oye, G.; Sjöblom, J.; Stöcker, M. Synthesis, characterization and potential applications of new materials in the mesoporous range. Adv. Colloid Interface Sci. 2001, 89–90, 439–466.
- Naono, H.; Hakuman, M.; Tsunehisa, T.; Tamura, N.; Nakai, K. Formation process of MCM-41 precursor and porous texture of MCM-41. J. Colloid Interface Sci. 2000, 224, 358–365.
- Huang, X.; Townley, H.E. An Assessment of mesoporous silica nanoparticle architectures as antigen carriers. Pharmaceutics 2020, 12, 294.
- Wang, S.; Wu, D.; Sun, Y.; Zhong, B. The synthesis of MCM-48 with high yields. Mater. Res. Bullet. 2001, 36, 1717–1720.
- Nandiyanto, A.B.D.; Kim, S.G.; Iskandar, F.; Okuyama, K. Synthesis of spherical mesoporous silica nanoparticles with nanometer-size controllable pores and outer diameters. Microporous Mesoporous Mater. 2009, 120, 447–453.
- Heikkilä, T.; Salonen, J.; Tuura, J.; Hamdy, M.S.; Mul, G.; Kumar, N.; Salmi, T.; Murzin, D.Y.; Laitinen, L.; Kaukonen, A.M.; et al. Mesoporous silica material TUD-1 as a drug delivery system. Int. J. Pharm. 2007, 331, 133–138.
- Kalbasi, J.R.; Zirakbash, A. Synthesis, characterization and drug release studies of poly(2-hydroxyethyl methacrylate)/KIT-5 nanocomposite as an innovative organic–inorganic hybrid carrier system. RSC Adv. 2015, 5, 12463–12471.
- Chircov, C.; Spoială, A.; Păun, C.; Cracium, L.; Ficai, D.; Ficai, A.; Andronescu, E.; Turcule, S.C. Mesoporous silica platforms with potential applications in release and adsorption of active agents. Molecules 2020, 25, 3814.
- Cai, Q.; Luo, Z.S.; Pang, W.Q.; Fan, Y.W.; Chen, X.H.; Cui, F.Z. Dilute solution routes to various controllable morphologies of MCM-41 silica with a basic medium. Chem. Mater. 2001, 13, 258–263.
- Fowler, C.E.; Khushalani, D.; Lebeau, B.; Mann, S. Nanoscale materials with mesostructured interiors. Adv. Mater. 2001, 13, 649–652.
- Nooney, R.I.; Thirunavukkarasu, D.; Chen, Y.M.; Josephs, R.; Ostafin, A.E. Synthesis of nanoscale mesoporous silica spheres with controlled particle size. Chem. Mater. 2002, 14, 4721–4728.
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459.
- Lin, Y.S.; Haynes, C.L. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J. Am. Chem. Soc. 2010, 132, 4834–4842.
- Williams, S.; Neumann, A.; Bremer, I.; Su, Y.; Dräger, G.; Kasper, C.; Behrens, P. Nanoporous silica nanoparticles as biomaterials: Evaluation of different strategies for the functionalization with polysialic acid by step-by-step cytocompatibility testing. J. Mater. Sci. Mater. Med. 2015, 26, 125.
- Qiao, Z.A.; Zhang, L.; Guo, M.; Liu, Y.; Huo, Q. Synthesis of mesoporous silica nanoparticles via controlled hydrolysis and condensation of silicon alkoxide. Chem. Mater. 2009, 21, 3823–3829.
- Blin, J.L.; Michaux, F.; Stébé, M.J. Nanostuctured mesoporous materials from different silica sources using fluorinated surfactants as templates. Colloids Surf. A Physicochem. Eng. Asp. 2016, 510, 104–112.
- Brevet, D.; Jouannin, C.; Tourné-Péteilh, C.; Devoisselle, J.M.; Vioux, A.; Viau, L. Self-encapsulation of a drug-containing ionic liquid into mesoporous silica monoliths or nanoparticles by a sol-gel process. RSC Adv. 2016, 6, 82916–82923.
- Siddiqui, B.; Rehman, A.; Haq, I.; Al-Dossary, A.A.; Elaissari, A.; Ahmed, N. Exploiting recent trends for the synthesis and surface functionalization of mesoporous silica nanoparticles towards biomedical applications. Int. J. Pharm. X 2022, 4, 10016.
- Wang, Y. Synthesis and formation of hierarchical mesoporous silica network in acidic aqueous solutions of sodium silicate and cationic surfactant. Colloid J. 2010, 72, 737–742.
- Das, D.; Yang, Y.; O’Brien, J.S.; Breznan, D.; Nimesh, S.; Bernatchez, S.; Hill, M.; Sayari, A.; Vincent, R.; Kumarathasan, P. Synthesis and physicochemical characterization of mesoporous SiO2 nanoparticles. J. Nanomater. 2014, 2014, 62.
- Li, Y.; Bastakoti, B.P.; Imura, M.; Tang, J.; Aldalbahi, A.; Torad, N.L.; Yamauchi, Y. Dual soft-template system based on colloidal chemistry for the synthesis of hollow mesoporous silica nanoparticles. Chem. Eur. J. 2015, 21, 6375–6380.
- Gai, F.; Zhou, T.; Chu, G.; Li, Y.; Liu, Y.; Huo, Q.; Akhtar, F. Mixed anionic surfactant-templated mesoporous silica nanoparticles for fluorescence detection of Fe3+. Dalton Trans. 2016, 45, 508–514.
- She, X.; Chen, L.; Velleman, L.; Li, C.; Zhu, H.; He, C.; Wang, T.; Shigdar, S.; Duan, W.; Kong, L. Fabrication of high specificity hollow mesoporous silica nanoparticles assisted by Eudragit for targeted drug delivery. J. Colloid Interface Sci. 2015, 445, 151–160.
- Um, K.; Chang, H.; Lee, K. Facile synthesis of hollow mesoporous zinc silicate nanoparticles using a dual surfactant system. RSC Adv. 2016, 6, 98717–98721.
- Napierska, D.; Thomassen, L.C.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39.
- Martin, K.R. The chemistry of silica and its potential health benefits. J. Nutr. Health Aging 2007, 11, 94–97.
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 2017, 29, 1604634.
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108.
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C. Donald Maxwell Parkin Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917.
- Malik, P.; Mukherjee, T.K. Recent Progress in gold and silver nanoparticles biomedical attributes towards lung and breast cancer treatment. Int. J. Pharm. 2018, 553, 483–509.
- Rizwanullah, M.; Ahmad, M.Z.; Ghoneim, M.M.; Alshehri, S.; Imam, S.S.; Md, S.; Alhakamy, N.A.; Jain, K.; Ahmad, J. Receptor-mediated targeted delivery of surface-modified nanomedicine in breast cancer: Recent update and challenges. Pharmaceutics 2021, 13, 2039.
- Duo, Y.; Li, Y.; Chen, C.; Liu, B.; Wang, X.; Zeng, X.; Chen, H. DOX-loaded pH-sensitive mesoporous silica nanoparticles coated with PDA and PEG induce pro-death autophagy in breast cancer. RSC Adv. 2017, 7, 39641–39650.
- Kumar, P.; Tambe, P.; Paknikar, K.M.; Gajbhiye, V. Folate/N-acetyl glucosamine conjugated mesoporous silica nanoparticles for targeting breast cancer cells: A comparative study. Colloids Surf. B 2017, 156, 203–212.
- Ren, W.; Iqbal, M.Z.; Zeng, L.; Chen, T.; Pan, Y.; Zhao, J.; Yin, H.; Zhang, L.; Zhang, J.; Li, A.; et al. Black TiO2 based core–shell nanocomposites as doxorubicin carriers for thermal imaging guided synergistic therapy of breast cancer. Nanoscale 2017, 9, 11195–11204.
- Shen, Y.; Li, M.; Liu, T.; Liu, J.; Xie, Y.; Zhang, J.; Xu, S.; Liu, H. A dual-functional HER2 aptamer-conjugated, pH-activated mesoporous silica nanocarrier-based drug delivery system provides in vitro synergistic cytotoxicity in HER2-positive breast cancer cells. Int. J. Nanomed. 2019, 14, 4029–4044.
- Ali, O.M.; Bekhit, A.A.; Khattab, S.N.; Helmy, M.W.; Abdel-Ghany, Y.S.; Teleb, M.; Elzoghby, A.O. Synthesis of lactoferrin mesoporous silica nanoparticles for pemetrexed/ellagic acid synergistic breast cancer therapy. Colloids Surf. B 2020, 188, 110824.
- Xu, P.; Yao, J.; Li, Z.; Wang, M.; Zhou, L.; Zhong, G.; Zheng, Y.; Li, N.; Zhai, Z.; Yang, S.; et al. Therapeutic effect of doxorubicin-chlorin E6-loaded mesoporous silica nanoparticles combined with ultrasound on triple-negative breast cancer. Int. J. Nanomed. 2020, 15, 2659–2668.
- Moodley, T.; Singh, M. Polymeric mesoporous silica nanoparticles for combination drug delivery in vitro. Biointerface Res. Appl. Chem. 2021, 11, 11905–11919.