 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rumiana Tzoneva | -- | 3962 | 2023-07-25 20:55:34 | | | |
| 2 | Peter Tang | + 1 word(s) | 3963 | 2023-07-26 03:26:17 | | |
Video Upload Options
Endothelial cells are constantly exposed to environmental stress factors that, above a certain threshold, trigger cellular senescence and apoptosis. The altered vascular function affects new vessel formation and endothelial fitness, contributing to the progression of age-related diseases.
1. Introduction
2. The Dual Nature of Cellular Senescence
3. Endothelial Senescence
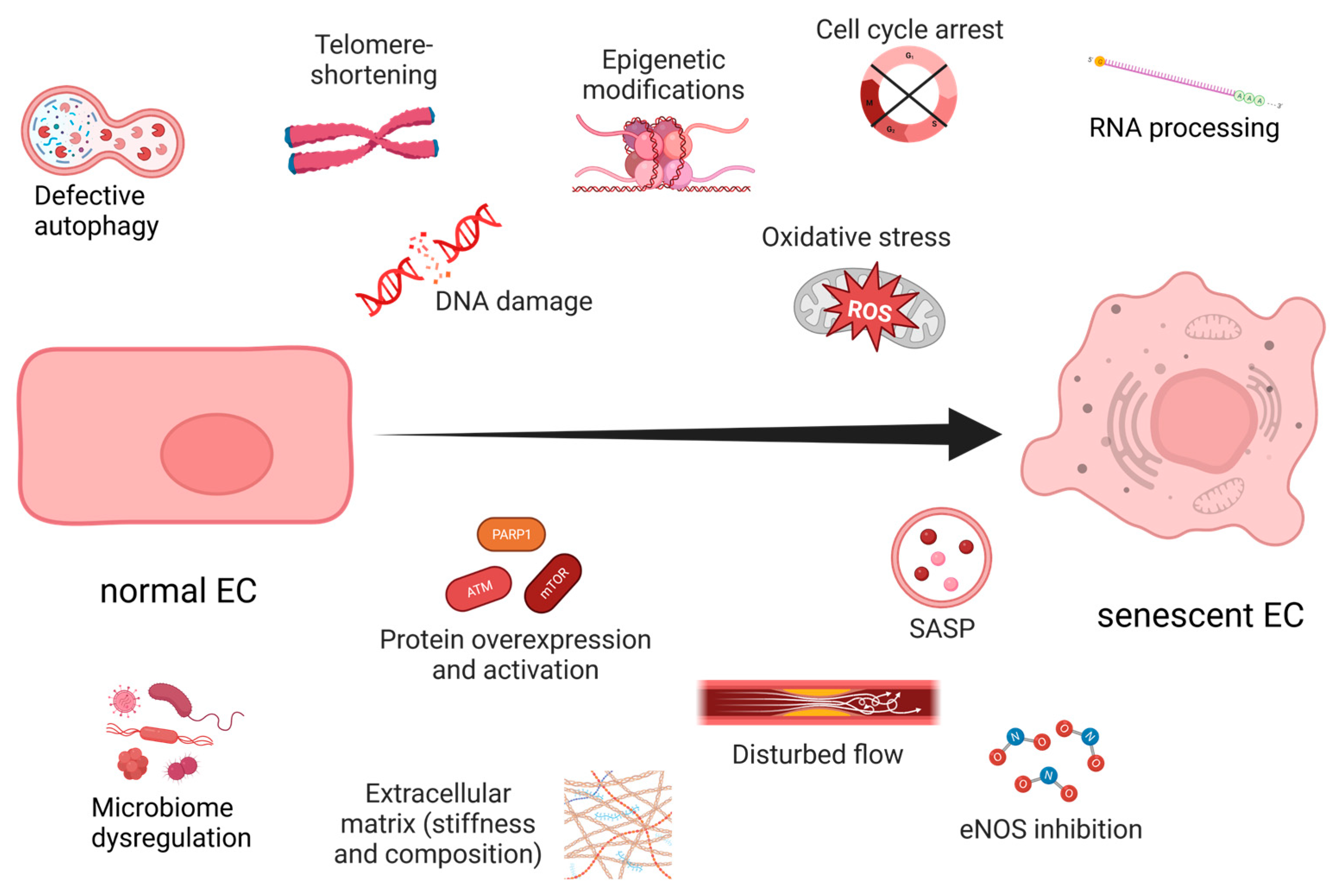
4. Unveiling the Interplay between Hypoxia and Oxidative Stress-Induced Endothelial Senescence
5. Exploring the Role of Extracellular Vesicles in Angiogenesis and Senescence
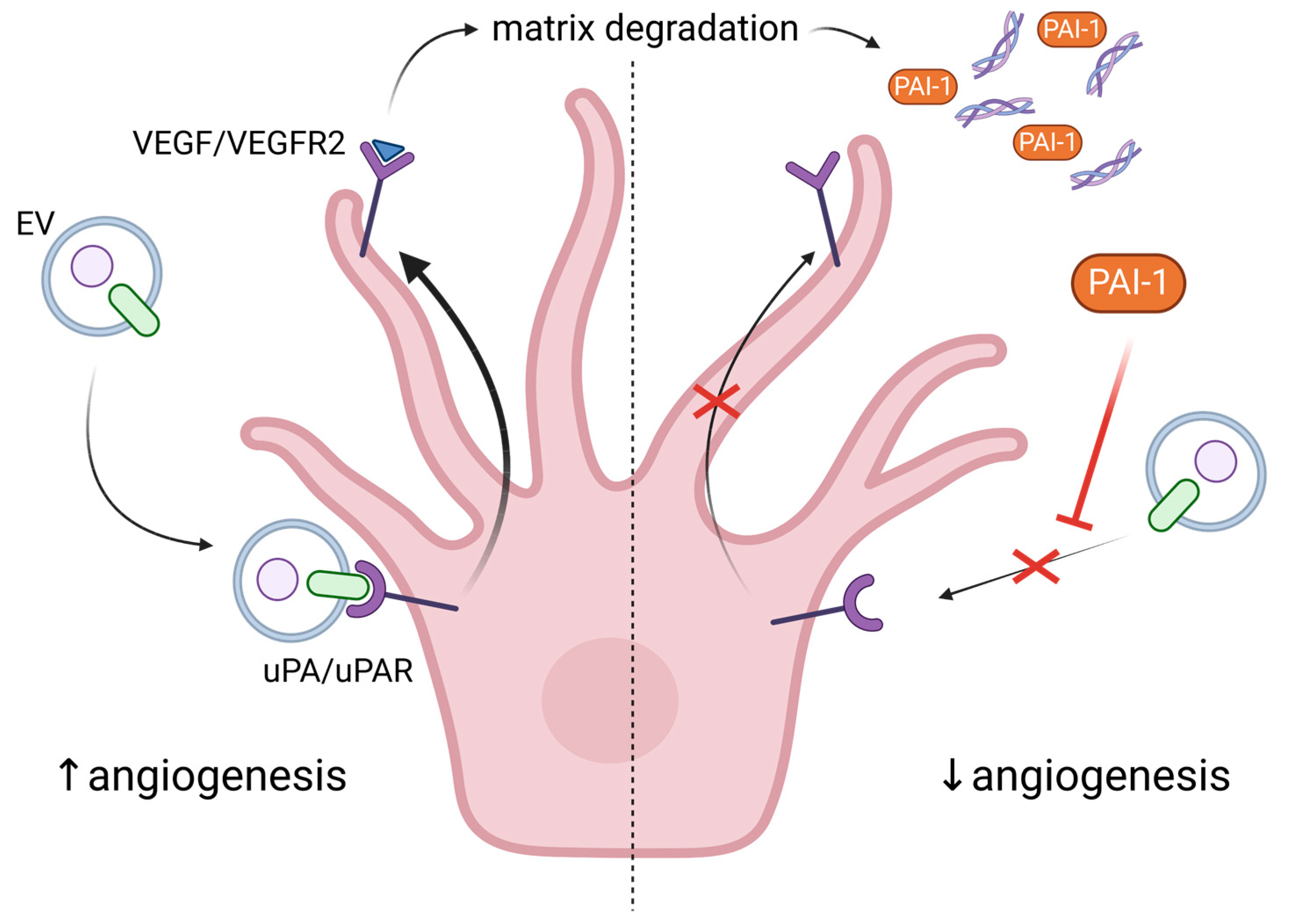
6. The Non-Productive Angiogenesis in Alzheimer’s Disease
|
AD Model |
Blood Vessels |
Protein Expression |
References |
|---|---|---|---|
|
Aβ monomers in HUVEC and zebrafish |
↑ capillary density |
- |
[75] |
|
Tau overexpressing mice; 15 months old |
↑ capillary density; ↑ angiogenesis; ↑ BBB permeability; ↓ CBF |
↑ VEGF; ↑ uPAR; ↑ MMP-9; ↑ PAI-1 |
[76] |
|
AD patients |
− |
↑ VEGF; ↑ TGF-β |
[77] |
|
HMVECs + Aβ monomers |
↓ angiogenesis |
↑ VEGFR1- ↑ senescence |
[78] |
|
APP-PSEN1/+ mice |
↑ non-productive angiogenesis; ↓ capillary density around plaques |
↑ VEGF |
[79] |
|
Tg2576 mice |
↓ capillary density around plaques |
↓ GLUT1 |
[80] |
|
AD patients; APP695 mice |
↓ capillary density |
VEGF supplementation improved cognitive function |
[81] |
|
3xTG-AD mice |
↑ capillary density; ↓ junction density |
− |
[82] |
7. Therapeutic Approaches to Endothelial Senescence and Dysfunction
7.1. Exercise Improves CBF, Vascular Function and Cognitive Performance
7.2. Caloric Restriction Reduces OS and Vascular Aging
7.3. Role of Resveratrol in the Vascular Biology and Senescence Process
References
- Patel-Hett, S.; D’Amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int. J. Dev. Biol. 2011, 55, 353–363.
- Adair, T.H.; Montani, J.-P. Overview of Angiogenesis. In Angiogenesis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010.
- Weinstein, N.; Mendoza, L.; Gitler, I.; Klapp, J. A network model to explore the effect of the micro-environment on endothelial cell behavior during angiogenesis. Front. Physiol. 2017, 8, 960.
- Brassard-Jollive, N.; Monnot, C.; Muller, L.; Germain, S. In vitro 3D Systems to Model Tumor Angiogenesis and Interactions With Stromal Cells. Front. Cell Dev. Biol. 2020, 8, 594903.
- Stryker, Z.I.; Rajabi, M.; Davis, P.J.; Mousa, S.A. Evaluation of angiogenesis assays. Biomedicines 2019, 7, 37.
- Lähteenvuo, J.; Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 2012, 110, 1252–1263.
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565.
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307.
- Ambrose, C.T. Pro-Angiogenesis Therapy and Aging: A Mini-Review. Gerontology 2017, 63, 393–400.
- Vinters, H.V.; Gilbert, J.J. Cerebral amyloid angiopathy: Incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983, 14, 924–928.
- Beckmann, N.; Schuler, A.; Mueggler, T.; Meyer, E.P.; Wiederhold, K.H.; Staufenbiel, M.; Krucker, T. Age-Dependent Cerebrovascular Abnormalities and Blood Flow Disturbances in APP23 Mice Modeling Alzheimer’s Disease. J. Neurosci. 2003, 23, 8453–8459.
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153.
- Singh, C.; Pfeifer, C.G.; Jefferies, W.A. Pathogenic Angiogenic Mechanisms in Alzheimer’s Disease. In Physiologic and Pathologic Angiogenesis-Signaling Mechanisms and Targeted Therapy; BoD–Books on Demand: Norderstedt, Germany, 2017.
- Bradaric, B.D.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for Angiogenesis in Parkinson’s disease, Incidental Lewy Body disease, and Progressive Supranuclear Palsy. J. Neural Transm. 2012, 119, 59.
- Ellison, S.M.; Trabalza, A.; Tisato, V.; Pazarentzos, E.; Lee, S.; Papadaki, V.; Goniotaki, D.; Morgan, S.; Mirzaei, N.; Mazarakis, N.D. Dose-dependent Neuroprotection of VEGF165 in Huntington’s DiseaseStriatum. Mol. Ther. 2013, 21, 1862.
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β oligomers in cellular models of Alzheimer’s disease. J. Neurochem. 2020, 155, 348–369.
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160.
- Jefferies, W.A.; Price, K.A.; Biron, K.E.; Fenninger, F.; Pfeifer, C.G.; Dickstein, D.L. Adjusting the compass: New insights into the role of angiogenesis in Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 64–69.
- Tchekalarova, J.; Tzoneva, R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. Int. J. Mol. Sci. 2023, 24, 3022.
- Tadokoro, K.; Ohta, Y.; Inufusa, H.; Loon, A.F.N.; Abe, K. Prevention of Cognitive Decline in Alzheimer’s Disease by Novel Antioxidative Supplements. Int. J. Mol. Sci. 2020, 21, 1974.
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nature 2005, 438, 954–959.
- Ribatti, D.; Guidolin, D. Morphogenesis of vascular and neuronal networks and the relationships between their remodeling processes. Brain Res. Bull. 2022, 186, 62–69.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194.
- Schmauck-Medina, T.; Molière, A.; Lautrup, S.; Zhang, J.; Chlopicki, S.; Madsen, H.B.; Cao, S.; Soendenbroe, C.; Mansell, E.; Vestergaard, M.B.; et al. New hallmarks of ageing: A 2022 Copenhagen ageing meeting summary. Aging 2022, 14, 6829–6839.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485.
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: Current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12.
- Ramos-Ibeas, P.; Gimeno, I.; Cañón-Beltrán, K.; Gutiérrez-Adán, A.; Rizos, D.; Gómez, E. Senescence and Apoptosis During in vitro Embryo Development in a Bovine Model. Front. Cell Dev. Biol. 2020, 8, 1646.
- De-Carvalho, D.P.; Jacinto, A.; Saúde, L. The right time for senescence. Elife 2021, 10, e72449.
- Wagner, M.; Hampel, B.; Bernhard, D.; Hala, M.; Zwerschke, W.; Jansen-Dürr, P. Replicative senescence of human endothelial cells in vitro involves G1 arrest, polyploidization and senescence-associated apoptosis. Exp. Gerontol. 2001, 36, 1327–1347.
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.C.; Gaugler, M.H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic. Biol. Med. 2017, 108, 750–759.
- Nakae, I.; Fujita, M.; Miwa, K.; Hasegawa, K.; Kihara, Y.; Nohara, R.; Miyamoto, S.; Ueda, K.; Tamaki, S.I.; Sasayama, S. Age-dependent impairment of coronary collateral development in humans. Heart Vessels 2000, 15, 176–180.
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of vascular aging. Circ. Res. 2018, 123, 849–867.
- Baffert, F.; Thurston, G.; Rochon-Duck, M.; Le, T.; Brekken, R.; McDonald, D.M. Age-Related Changes in Vascular Endothelial Growth Factor Dependency and Angiopoietin-1-Induced Plasticity of Adult Blood Vessels. Circ. Res. 2004, 94, 984–992.
- Lecce, L.; Lam, Y.T.; Lindsay, L.A.; Yuen, S.C.; Simpson, P.J.L.; Handelsman, D.J.; Ng, M.K.C. Aging Impairs VEGF-Mediated, Androgen-Dependent Regulation of Angiogenesis. Mol. Endocrinol. 2014, 28, 1487–1501.
- Welford, S.M.; Giaccia, A.J. Hypoxia and Senescence: The impact of oxygenation on tumor suppression. Mol. Cancer Res. 2011, 9, 538.
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 2007, 100, 1659–1666.
- Yang, W.; Hekimi, S. A Mitochondrial Superoxide Signal Triggers Increased Longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, 1000556.
- Ungvari, Z.; Tucsek, Z.; Sosnowska, D.; Toth, P.; Gautam, T.; Podlutsky, A.; Csiszar, A.; Losonczy, G.; Valcarcel-Ares, M.N.; Sonntag, W.E.; et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 877–891.
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 2004, 117, 2417–2426.
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742.
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A ROS-dependent mechanism promotes CDK2 phosphorylation to drive progression through S phase. Dev. Cell 2022, 57, 1712–1727.e9.
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Conboy, 2005, Nature, Rejuvenecimento celular e nicho.pdf. Nature 2005, 433, 760–764.
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663.
- Jeon, O.H.; Mehdipour, M.; Gil, T.H.; Kang, M.; Aguirre, N.W.; Robinson, Z.R.; Kato, C.; Etienne, J.; Lee, H.G.; Alimirah, F.; et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat. Metab. 2022, 4, 995–1006.
- Oh, C.; Koh, D.; Jeon, H.B.; Kim, K.M. The Role of Extracellular Vesicles in Senescence. Mol. Cells 2022, 45, 603–609.
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular Vesicles in Angiogenesis. Circ. Res. 2017, 120, 1658–1673.
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228.
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383.
- Kuriyama, N.; Yoshioka, Y.; Kikuchi, S.; Azuma, N.; Ochiya, T. Extracellular Vesicles Are Key Regulators of Tumor Neovasculature. Front. Cell Dev. Biol. 2020, 8, 611039.
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840.
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287.
- Riquelme, J.A.; Takov, K.; Santiago-Fernández, C.; Rossello, X.; Lavandero, S.; Yellon, D.M.; Davidson, S.M. Increased production of functional small extracellular vesicles in senescent endothelial cells. J. Cell. Mol. Med. 2020, 24, 4871–4876.
- Choi, E.J.; Kil, I.S.; Cho, E.G. Extracellular Vesicles Derived from Senescent Fibroblasts Attenuate the Dermal Effect on Keratinocyte Differentiation. Int. J. Mol. Sci. 2020, 21, 1022.
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e6.
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002, 160, 673–680.
- Alexander, R.A.; Prager, G.W.; Mihaly-Bison, J.; Uhrin, P.; Sunzenauer, S.; Binder, B.R.; Schütz, G.J.; Freissmuth, M.; Breuss, J.M. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc. Res. 2012, 94, 125–135.
- Breuss, J.M.; Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adh. Migr. 2012, 6, 535.
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.Q.N.; et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117.
- Brunner, P.M.; Heier, P.C.; Mihaly-Bison, J.; Priglinger, U.; Binder, B.R.; Prager, G.W. Density enhanced phosphatase-1 down-regulates urokinase receptor surface expression in confluent endothelial cells. Blood 2011, 117, 4154–4161.
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. PAI-1 is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446.
- van Balkom, B.W.M.; de Jong, O.G.; Smits, M.; Brummelman, J.; den Ouden, K.; de Bree, P.M.; van Eijndhoven, M.A.J.; Pegtel, D.M.; Stoorvogel, W.; Würdinger, T.; et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013, 121, 3997–4006.
- Ramakrishnan, D.P.; Hajj-Ali, R.A.; Chen, Y.; Silverstein, R.L. Extracellular vesicles activate a CD36-dependent signaling pathway to inhibit microvascular endothelial cell migration and tube formation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 534–544.
- Mezentsev, A.; Merks, R.M.H.; O’Riordan, E.; Chen, J.; Mendelev, N.; Goligorsky, M.S.; Brodsky, S.V. Endothelial microparticles affect angiogenesis in vitro: Role of oxidative stress. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, 1106–1114.
- Lu, Q.; Qin, H.; Tan, H.; Wei, C.; Yang, X.; He, J.; Liang, W.; Li, J. Senescence Osteoblast-Derived Exosome-Mediated miR-139-5p Regulates Endothelial Cell Functions. Biomed Res. Int. 2021, 2021, 5576023.
- Wong, P.F.; Tong, K.L.; Jamal, J.; Khor, E.S.; Lai, S.L.; Mustafa, M.R. Senescent HUVECs-secreted exosomes trigger endothelial barrier dysfunction in young endothelial cells. Excli J. 2019, 18, 764–776.
- Lacroix, R.; Sabatier, F.; Mialhe, A.; Basire, A.; Pannell, R.; Borghi, H.; Robert, S.; Lamy, E.; Plawinski, L.; Camoin-Jau, L.; et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: A mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood 2007, 110, 2432–2439.
- Ou, Z.J.; Chang, F.J.; Luo, D.; Liao, X.L.; Wang, Z.P.; Zhang, X.; Xu, Y.Q.; Ou, J.S. Endothelium-derived microparticles inhibit angiogenesis in the heart and enhance the inhibitory effects of hypercholesterolemia on angiogenesis. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 661–668.
- Li, Y.; Bax, C.; Patel, J.; Vazquez, T.; Ravishankar, A.; Bashir, M.M.; Grinnell, M.; Diaz, D.; Werth, V.P. Plasma-derived DNA containing-extracellular vesicles induce STING-mediated proinflammatory responses in dermatomyositis. Theranostics 2021, 11, 7144–7158.
- Yu, H.; Liao, K.; Hu, Y.; Lv, D.; Luo, M.; Liu, Q.; Huang, L.; Luo, S. Role of the cGAS-STING Pathway in Aging-related Endothelial Dysfunction. Aging Dis. 2022, 13, 1901.
- Park, J.H.; Hong, J.H.; Lee, S.W.; Ji, H.D.; Jung, J.A.; Yoon, K.W.; Lee, J.I.; Won, K.S.; Song, B.I.; Kim, H.W. The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: A positron emission tomography study in rats. Sci. Rep. 2019, 9, 14102.
- Qiu, L.; Ng, G.; Tan, E.K.; Liao, P.; Kandiah, N.; Zeng, L. Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci. Rep. 2016, 6, 23964.
- Babcock, K.R.; Page, J.S.; Fallon, J.R.; Webb, A.E. Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Rep. 2021, 16, 681–693.
- Adamovich, Y.; Ladeuix, B.; Golik, M.; Koeners, M.P.; Asher, G. Rhythmic Oxygen Levels Reset Circadian Clocks through HIF1α. Cell Metab. 2017, 25, 93–101.
- Cameron, D.J.; Galvin, C.; Alkam, T.; Sidhu, H.; Ellison, J.; Luna, S.; Ethell, D.W. Alzheimer’s-Related Peptide Amyloid-b Plays a Conserved Role in Angiogenesis. PLoS ONE 2012, 7, e39598.
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298.
- Alvarez-Vergara, M.I.; Rosales-Nieves, A.E.; March-Diaz, R.; Rodriguez-Perinan, G.; Lara-Ureña, N.; Ortega-de San Luis, C.; Sanchez-Garcia, M.A.; Martin-Bornez, M.; Gómez-Gálvez, P.; Vicente-Munuera, P.; et al. Non-productive angiogenesis disassembles Aß plaque-associated blood vessels. Nat. Commun. 2021, 12, 3098.
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243.
- Angom, R.S.; Wang, Y.; Wang, E.; Pal, K.; Bhattacharya, S.; Watzlawik, J.O.; Rosenberry, T.L.; Das, P.; Mukhopadhyay, D. VEGF receptor-1 modulates amyloid β 1-42 oligomer-induced senescence in brain endothelial cells. FASEB J. 2019, 33, 4626.
- Kouznetsova, E.; Klingner, M.; Sorger, D.; Sabri, O.; Großmann, U.; Steinbach, J.; Scheunemann, M.; Schliebs, R. Developmental and amyloid plaque-related changes in cerebral cortical capillaries in transgenic Tg2576 Alzheimer mice. Int. J. Dev. Neurosci. 2006, 24, 187–193.
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, srep02053.
- Jullienne, A.; Quan, R.; Szu, J.I.; Trinh, M.V.; Behringer, E.J.; Obenaus, A. Progressive Vascular Abnormalities in the Aging 3xTg-AD Mouse Model of Alzheimer’s Disease. Biomedicines 2022, 10, 1967.
- Steinman, J.; Sun, H.S.; Feng, Z.P. Microvascular Alterations in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 14, 472.
- Rahbarghazi, A.; Siahkouhian, M.; Rahbarghazi, R.; Ahmadi, M.; Bolboli, L.; Keyhanmanesh, R.; Mahdipour, M.; Rajabi, H. Role of melatonin in the angiogenesis potential; highlights on the cardiovascular disease. J. Inflamm. 2021, 18, 4.
- Goradel, N.H.; Asghari, M.H.; Moloudizargari, M.; Negahdari, B.; Haghi-Aminjan, H.; Abdollahi, M. Melatonin as an angiogenesis inhibitor to combat cancer: Mechanistic evidence. Toxicol. Appl. Pharmacol. 2017, 335, 56–63.
- Viboolvorakul, S.; Patumraj, S. Exercise training could improve age-related changes in cerebral blood flow and capillary vascularity through the upregulation of VEGF and eNOS. Biomed Res. Int. 2014, 2014, 230791.
- Soto, I.; Graham, L.C.; Richter, H.J.; Simeone, S.N.; Radell, J.E.; Grabowska, W. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015, 13, 1002279.
- Archer, T. Physical exercise alleviates debilities of normal aging and Alzheimer Õ s disease. Acta Neurol. Scand. 2011, 123, 221–238.
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557.
- Ding, Y.; Li, J.; Zhou, Y.; Rafols, J.A.; Clark, J.C.; Ding, Y. Cerebral Angiogenesis and Expression of Angiogenic Factors in Aging Rats after Exercise. Curr. Neurovascular Res. 2006, 3, 15–23.
- Frühbeis, C.; Helmig, S.; Tug, S.; Simon, P.; Krämer-Albers, E.M. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J. Extracell. Vesicles 2015, 4, 28239.
- Brahmer, A.; Neuberger, E.; Esch-Heisser, L.; Haller, N.; Jorgensen, M.M.; Baek, R.; Möbius, W.; Simon, P.; Krämer-Albers, E.M. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J. Extracell. Vesicles 2019, 8, 1615820.
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A.; Mccarthy, J.J. Extracellular Vesicles and Exosomes: Insights From Exercise Science. Front. Physiol. 2021, 11, 604274.
- Boerman, E.M.; Everhart, J.E.; Segal, S.S. Advanced age decreases local calcium signaling in endothelium of mouse mesenteric arteries in vivo. Am. J. Physiol.-Heart Circ. Physiol. 2016, 310, H1091–H1096.
- Kumar, V.B.S.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial cell response to lactate: Implication of PAR modification of VEGF. J. Cell. Physiol. 2007, 211, 477–485.
- Porporato, P.E.; Payen, V.L.; De Saedeleer, C.J.; Préat, V.; Thissen, J.P.; Feron, O.; Sonveaux, P. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis 2012, 15, 581–592.
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719.
- Ewald, C.Y.; Castillo-Quan, J.I.; Blackwell, T.K. Untangling longevity, dauer, and healthspan in Caenorhabditis elegans insulin/IGF-1-signalling. Gerontology 2018, 64, 96.
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.O.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430.
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.P.; Ming, X.F.; Yang, Z. Hyperactive S6K1 Mediates Oxidative Stress and Endothelial Dysfunction in Aging: Inhibition by Resveratrol. PLoS ONE 2011, 6, e19237.
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-Long Caloric Restriction Reduces Oxidative Stress and Preserves Nitric Oxide Bioavailability and Function in Arteries of Old Mice. Aging Cell 2013, 12, 772.
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650.
- Martens, C.R.; Seals, D.R. Practical alternatives to chronic caloric restriction for optimizing vascular function with ageing. J. Physiol. 2016, 594, 7177–7195.
- Custodia, A.; Ouro, A.; Romaus-Sanjurjo, D.; Pías-Peleteiro, J.M.; de Vries, H.E.; Castillo, J.; Sobrino, T. Endothelial Progenitor Cells and Vascular Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 13, 811210.
- Liao, N.; Shi, Y.; Zhang, C.; Zheng, Y.; Wang, Y.; Zhao, B.; Zeng, Y.; Liu, X.; Liu, J. Antioxidants inhibit cell senescence and preserve stemness of adipose tissue-derived stem cells by reducing ROS generation during long-term in vitro expansion. Stem Cell Res. Ther. 2019, 10, 306.
- Sovernigo, T.C.; Adona, P.R.; Monzani, P.S.; Guemra, S.; Barros, F.D.A.; Lopes, F.G.; Leal, C.L.V. Effects of supplementation of medium with different antioxidants during in vitro maturation of bovine oocytes on subsequent embryo production. Reprod. Domest. Anim. 2017, 52, 561–569.
- Guillot, E.; Lemay, A.; Allouche, M.; Vitorino Silva, S.; Coppola, H.; Sabatier, F.; Dignat-George, F.; Sarre, A.; Peyter, A.C.; Simoncini, S.; et al. Resveratrol Reverses Endothelial Colony-Forming Cell Dysfunction in Adulthood in a Rat Model of Intrauterine Growth Restriction. Int. J. Mol. Sci. 2023, 24, 9747.

