Endothelial cells are constantly exposed to environmental stress factors that, above a certain threshold, trigger cellular senescence and apoptosis. The altered vascular function affects new vessel formation and endothelial fitness, contributing to the progression of age-related diseases.
- angiogenesis
- cellular senescence
- aging
- extracellular vesicles
- oxidative stress
- Alzheimer’s disease
1. Introduction
2. The Dual Nature of Cellular Senescence
Cellular senescence is a fundamental process associated with tissue homeostasis during development, first described by Hayflick and Moorhead [23]. The authors observed a terminal pause in cell division of normal human fibroblasts after several cycles of passaging. They concluded that cultured cells cease to proliferate upon a finite number of doublings and, therefore, could be used as a model for aging. Today, this is referred to as the Hayflick limit. The processes of senescence and aging are intertwined in the sense that aging progresses with time and associates with increased numbers of senescent cells. Therefore, cellular senescence is also accepted as a hallmark of aging and a risk factor for age-related neurodegenerative diseases. However, senescence occurs during the full lifespan of an individual and is not restricted to later life stages. The resulting inability to divide is a consequence of irreversible cell cycle arrest, caused by the accumulation of various stress factors such as DNA damage, inflammation, telomere shortening, chromatin perturbations, and oncogene induction [12][24][25][26][12,24,25,26]. Senescence is believed to have evolved as a protective mechanism against cancer, but it also contributes to age-related physiological decline [27]. Additionally, loss of senescence during embryonic development allows the progression of unhealthy cells in embryos [28]. In contrast, while protecting against the propagation of mutated DNA, senescence harms long-living organisms, as it inhibits tissue renewal and function. These observations gave rise to the idea that there is a “right time to senesce”, arguing that the end goal of the fight against aging is not to completely eliminate senescent cells (SCs) but to learn how to tame them [29].3. Endothelial Senescence
Aging and prolonged exposure to environmental factors, such as toxins, ROS, shear stress, and extracellular matrix (ECM) perturbations, induce senescence in ECs (Figure 1). Interestingly, unlike most SCs, senescent ECs (sen-ECs) remain susceptible to apoptosis [30][63], a mechanism most likely evolved to rearrange the microvasculature and counteract proliferation. Senescence in ECs is usually triggered by telomere shortening [26], which can be avoided by the exogenous introduction of telomerase [6]. Ionizing radiation can also geroconvert human microvascular cells in a time- and dose-dependent manner, predominantly by uncoupling Complex II of the mitochondrial respiratory chain [31][64], demonstrating ECs’ susceptibility to OS. In any case, the balance between senescence and angiogenesis becomes dysregulated during aging and neurodegenerative diseases, but the underlying mechanisms remain elusive. The negative consequences of vascular aging are apparent in older people in whom the regeneration of blood flow after ischemia or wounding is a slow and tedious process [32][65]. The accumulated stress over time reduces the proliferative capacity of ECs and modifies their interaction with the already altered ECM [33][66]. Furthermore, aging reduces the general expression of vascular endothelial growth factor (VEGF) [6] and promotes angiogenic incompetence in ECs, making them unable to respond to VEGF [7]. Some of the suggested reasons for the VEGF insensitivity are the age-related loss of VEGF receptor 2 (VEGFR2) [34][67], androgen resistance [35][68] and reduction in nitric oxide (NO) [6]. Furthermore, the SASP can directly inhibit angiogenesis by secreting factors that block endothelial cell proliferation and migration. At the same time, SCs can induce angiogenesis by secreting pro-inflammatory cytokines that promote neovascularization.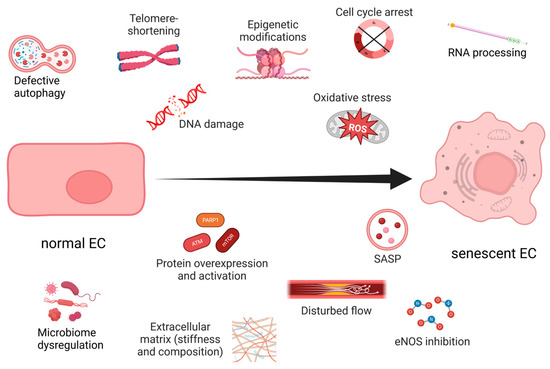
4. Unveiling the Interplay between Hypoxia and Oxidative Stress-Induced Endothelial Senescence
5. Exploring the Role of Extracellular Vesicles in Angiogenesis and Senescence
Legends about the infamous Hungarian Countess Elizabeth Bathory tell the story of her supposed anti-aging process of bathing in the blood of young girls. A similar idea governs the myths for vampires, which might not necessarily stay young, but become immortal by feeding on human blood. Surprisingly, there seems to be some truth in these myths, as recent studies showed that blood exchange from young to old mice rejuvenates them, but the opposite transfusion leads to senescence in the young [43][44][45][121,122,123]. The latter highlights the role of SASP in aging, which assists the immune response and, in the context of angiogenesis, influences new vessel formation. In addition to soluble factors, such as chemokines, inflammatory cytokines and growth factors, extracellular vesicles (EVs) are key components of SASP (reviewed in [46][124]). EVs are a very heterogeneous group of membranous structures, roughly categorized into three main groups based on size and origin: apoptotic bodies (ABs), microvesicles (MVs) that range from 50 to 5000 nm and are formed by outward budding and fission of the plasma membrane, and exosomes (30–100 nm) that are produced by the fusion of multivesicular endosomes with the plasma membrane, releasing intraluminal vesicles into the extracellular space. The EVs play important roles in intercellular communication, and their release is a strictly regulated process [47][125]. They are involved in both physiological and pathological processes and play a role in intercellular communication through the transfer of proteins, lipids, and nucleic acids [48][49][126,127]. EVs are implicated in cancer etiology due to their ability to promote cancer cell migration, transformation of non-malignant cells and pro-angiogenic activity [50][128]. While healthy cells release EVs as part of normal cellular homeostasis, senescent cells secrete EVs that have a significant role in angiogenesis and neurodegenerative disease progression. The presence of pro-angiogenic molecules like HIF-1α, VEGF, MMPs, and microRNAs in EVs [51][129] may lead to homeostasis disruption and non-productive angiogenesis. The role of EVs as key functional components of SASP is further highlighted by the observation that secretion of EVs is much higher in different types of senescent cells, including ECs, as compared to young ones [52][53][130,131]. A possible explanation for this is the observed upregulation of neutral sphingomyelinase and dysfunction of lysosomal activity in senescent cells [54][132]. One study even suggests that hypoxia prevents senescence by decreasing the SASP, rather than reducing the number of senescent cells [55][118]. The important role of EVs from ECs, as well as other blood cell types, in angiogenesis is summarized here [47][125]. More specifically, EVs from ECs are rich in β1 integrins and metalloproteinases (MMP-2 and MMP-9), which allow them to penetrate the ECM, to remodel it and to form tubular capillary-like structures. Stimulation with VEGF and FGF-2 facilitates the association of the active and proenzyme forms of the MMPs with EC-derived vesicles [56][133]. EVs can also transport urokinase plasminogen activator/uPA receptor (uPA/uPAR), which are both pro-angiogenic. It was shown that uPAR modulates VEGF-induced EC migration by balancing the proteolysis of the ECM and the cell motility through integrin-associated focal adhesion (Figure 2). Revu Ann Alexander and colleagues demonstrated that VEGF causes endocytosis of αVβI integrin and activation of uPA/uPAR, resulting in matrix degradation [57][134]. Another active participant in this process is the inhibitor of uPA—plasminogen activator inhibitor (PAI-1), which is released from the degraded matrix and internalized, further directing the balance toward invasive cell migration, i.e., angiogenesis (Figure 2). Inhibition or deficiency of uPAR suppressed VEGF-induced angiogenesis in tumor cells [58][135] or in mice [59][136], respectively. Moreover, uPAR stimulated angiogenesis through VEGFR2, which upon internalization activates other pro-angiogenic stimuli [59][136]. In confluent ECs, the expression of uPAR is down-regulated compared to sub-confluent proliferative cells, thus preventing VEGF-activated signaling and angiogenesis [60][137]. In addition, levels of PAI-1 are elevated in senescent and aged ECs, making it a useful marker for senescence [61][138]. Besides inhibiting uPAR, PAI-1 also induces p53 and p21, activity that is suppressed by SIRT1 overexpression in endothelial cells. SIRT1 is also able to induce eNOS activity, protecting ECs from endothelial dysfunction [61][138]. The pro-angiogenic properties of exosomes from ECs may also be attributed to EV-associated micro RNAs such as miR-214 [62][139]. More specifically, the latter prevents senescence through silencing ATM in recipient cells.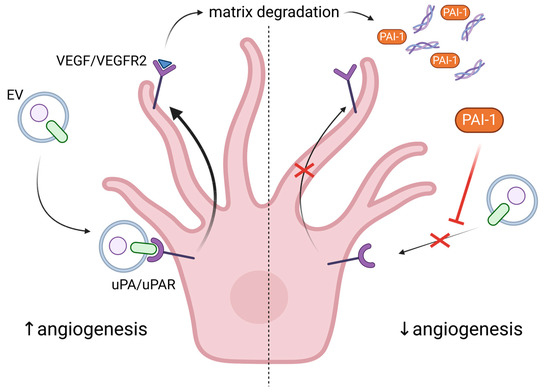
6. The Non-Productive Angiogenesis in Alzheimer’s Disease
Currently, there are two main hypotheses for the development of AD—the accumulation of amyloid plaques (Aβ) due to an error in the metabolism of the amyloid precursor protein (APP); and the hyperphosphorylation of Tau (or p-Tau), resulting in microtubule polymerization catastrophe and formation of fibrils [16]. APP is a transmembrane glycoprotein separated into an intracellular C-terminal, Aβ transmembrane and N-terminal extracellular domains. Its primary function is interneuronal communication, and once it performs it, APP is degraded by α- and γ-secretase to a soluble, non-amyloid form, or by β- and γ-secretase to insoluble Aβ1–40 and Aβ1–42 isoforms [16]. In animal models, elevated levels of Aβ1–42 and p-Tau were correlated with cerebrovascular dysfunction, chronic hypoperfusion and worsened AD symptoms [71][72][148,149]. One of the most affected brain areas in AD is the hippocampus, which is normally able to continue with adult neurogenesis. Thus, a decline in neurogenesis could be used as a marker for AD progression in animal models [73][150]. In fact, thwe researchers demonstrated worsened long-term memory and anxiety in a rat model of icvAβ1–42 concomitant with pinealectomy (AD with melatonin deficiency). These behaviors are controlled by the hippocampus and corresponded with increased OS in the structure [74][108]. Pro-inflammatory cytokines, such as interleukin-1β, become abundant during AD and induce the expression of VEGF, yielding new blood vessels [18]. Although angiogenesis is initiated around Aβ plaques, the process is non-productive, leading to the disassembly of Aβ plaque-associated blood vessels and the phagocytic activity of microglia [75][151]. However, there is conflicting evidence relating the cause of AD and whether there is an increase or decrease in blood vessel density [75][76][77][78][79][80][81][82][151,152,153,154,155,156,157,158] (Table 1).AD Model | Blood Vessels | Protein Expression | References | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Aβ monomers in HUVEC and zebrafish | ↑ capillary density | - |
[75] |
[151] |
||||||||||||||||||
Tau overexpressing mice; 15 months old | ↑ capillary density; | ↑ angiogenesis; | ↑ BBB permeability; | ↓ CBF | ↑ VEGF; | ↑ uPAR; | ↑ MMP-9; | ↑ PAI-1 |
[76] |
[152] |
||||||||||||
AD patients | − | ↑ VEGF; | ↑ TGF-β |
[77] |
[153] |
|||||||||||||||||
HMVECs + Aβ monomers | ↓ angiogenesis | ↑ VEGFR1- | ↑ senescence |
[78] |
[154] |
|||||||||||||||||
APP-PSEN1/+ mice | ↑ non-productive angiogenesis; | ↓ capillary density around plaques | ↑ VEGF |
[79] |
[155] |
|||||||||||||||||
Tg2576 mice | ↓ capillary density around plaques | ↓ GLUT1 |
[80] |
[156] |
||||||||||||||||||
AD patients; APP695 mice | ↓ capillary density | VEGF supplementation improved cognitive function |
[81] |
[157] |
||||||||||||||||||
3xTG-AD mice | ↑ capillary density; | ↓ junction density | − |
[82] |
[158] |