Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Partha Pratim Manna | -- | 1745 | 2023-05-22 06:46:30 | | | |
| 2 | Dean Liu | -2 word(s) | 1743 | 2023-05-24 04:23:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Singh, R.; Kashif, M.; Srivastava, P.; Manna, P.P. Chemotherapeutics for Leishmaniasis. Encyclopedia. Available online: https://encyclopedia.pub/entry/44626 (accessed on 23 July 2026).
Singh R, Kashif M, Srivastava P, Manna PP. Chemotherapeutics for Leishmaniasis. Encyclopedia. Available at: https://encyclopedia.pub/entry/44626. Accessed July 23, 2026.
Singh, Ranjeet, Mohammad Kashif, Prateek Srivastava, Partha Pratim Manna. "Chemotherapeutics for Leishmaniasis" Encyclopedia, https://encyclopedia.pub/entry/44626 (accessed July 23, 2026).
Singh, R., Kashif, M., Srivastava, P., & Manna, P.P. (2023, May 22). Chemotherapeutics for Leishmaniasis. In Encyclopedia. https://encyclopedia.pub/entry/44626
Singh, Ranjeet, et al. "Chemotherapeutics for Leishmaniasis." Encyclopedia. Web. 22 May, 2023.
Copy Citation
Leishmaniasis, a category 1 neglected protozoan disease caused by a kinetoplastid pathogen called Leishmania, is transmitted through dipteran insect vectors (phlebotomine, sand flies) in three main clinical forms: fatal visceral leishmaniasis, self-healing cutaneous leishmaniasis, and mucocutaneous leishmaniasis.
chemoinformatics
bioinformatics
drugs
inhibitors
1. Introduction
Leishmaniasis is a group of vector-borne infectious protozoan diseases endemic to nearly one hundred countries [1]. Leishmaniasis is considered by the WHO to be a neglected tropical disease and a major international health challenge. In addition to malaria, it is the deadliest parasitic disease worldwide. Nearly 0.71–1 million new cases and approximately 20,000 to 65,000 deaths are reported annually, predominantly in socioeconomically vulnerable communities with limited access to essential medicines. Over 20 different species of Leishmania are reported to be infective to humans, categorized as Old World (Mediterranean countries, Asia, and Africa) and New World (America) forms. The dipteran fly Phlebotomus and its subspecies in the Old World and Lutzomyia in the New World are proven vectors for human leishmaniasis. The disease globally affects approximately 14 million people, with over one billion people at high risk of infection [2]. A World Health Organization (WHO)-sponsored epidemiological report indicates that there are nearly 12 million active cases of leishmaniasis. The incidence of cutaneous leishmaniasis is two to three times more common than visceral leishmaniasis [3][4]. At present, this disease results in 20,000 to 65,000 deaths reported annually and is included among the 18 most neglected tropical diseases (NTDs). Approximately one hundred species of these dipteran insects belonging to the genera Phlebotomus and Lutzomyia are known as the main vectors involved in biological transmission [4]. Leishmania has a complex life cycle characterized by the presence of digenetic stages: flagellated promastigotes and flagellated amastigotes [5]. The metacyclic promastigote form in sand flies is responsible for infection in healthy individuals. The amastigote form is known for its pathogenesis, having a spherical shape with a rudimentary flagellum. Amastigotes reside, propagate, and persist within the host’s mononuclear phagocytic cells [6]. Leishmaniasis represents a wide spectrum pathology ranging from less severe and self-curable cutaneous leishmaniasis (CL) to more severe and fatal visceral leishmaniasis (VL). The clinical symptoms due to parasitic infections are classified into three types of disease: cutaneous, mucocutaneous, and visceral leishmaniasis [5][6]. Cutaneous leishmaniasis is manifested by the development of skin lesions and is the most common type prevalent in the Middle East. Visceral leishmaniasis, on the other hand, is distinguished by the occurrence of hepatosplenomegaly, fever, and weight loss, and is considered as a serious health hazard for the infected individual. Mucocutaneous leishmaniasis (MCL) is characterized by damage to oral mucous membranes in the nose, mouth, and throat, which potentiates inflammation and face disfiguration [7]. Recently, a new subgenus, Mundinia, has been reported, and a member of this group (L. martiniquensis) causes VL in Southeast Asian regions. L. martiniquensis typically causes VL in humans and can be treated with amphotericin B as a first-line chemotherapeutic option. It has been reported that VL caused by L. martiniquensis has a higher relapse rate and occurs in individuals with HIV infection [8][9].
In addition to two other kinetoplastid pathogens, viz. for Trypanosoma cruzi and Trypanosoma brucei, the management of leishmaniasis requires integrated and multidisciplinary strategies that include vector control, enhanced diagnostics, and increased awareness of new therapies with safe and efficient medicines [10]. There is still no effective vaccine available, and the control of the disease primarily rests on chemotherapy, the majority of which is costly and has a wide array of side effects [10][11]. Pentavalent antimonials (sodium stibogluconate, meglumine antimoniate or generic formulations) have been used as standard drugs in countries such as India and Nepal for over 60 years and remain the primary treatment options in many endemic regions despite widespread parasite resistance [12][13]. A single dose of the polyene antibiotic amphotericin B demonstrated a 95% efficiency against visceral leishmaniasis in India [14]. Intravenous administration of liposomal amphotericin B has become a standard treatment in many countries but remains expensive, even for single-course treatments [15][16]. Miltefosine, an alkyl-lysophospholipid analog, was initially developed as an anticancer compound and is considered as a first-line effective oral drug against Leishmania [17][18]. Miltefosine has been used successfully for the treatment of VL in India since 2002 and has been incorporated into the visceral leishmaniasis elimination program for the Indian subcontinent [19]. Despite some success, miltefosine administration registers considerable resistance, with relapse in nearly 20% of patients post-treatment [20]. Miltefosine was also found to be effective against cutaneous and mucocutaneous leishmaniasis in South America, with considerable differences in percent success in therapy programs [21][22][23]. The aminoglycoside paromomycin has been approved for the treatment of VL. The efficacy of paromomycin against VL patients has been demonstrated in phase III clinical trials in India [24][25]. However, paromomycin has also shown shortcomings in efficacy across geographical regions, as demonstrated by its less than satisfactory trial in Sudan compared to India [26][27]. It is known that the drugs used act via different cellular and molecular mechanisms, causing a variety of outcomes including the apoptosis of parasites, but are also widely associated with variable toxicity and setbacks in sought-after results. These difficulties are further complicated by the emergence of drug resistance against parasites across the globe that have persisted alongside conventional chemotherapy practiced in endemic areas. This significantly lowers the susceptibility to drugs and the emergence of difficult-to-treat resistant variants of the same species [28][29][30].
Many investigators have highlighted the need to discover new drug targets employing the knowledge of parasite biochemistry to develop revolutionary new drugs by using emerging technologies. Several natural and synthetic drugs as well as repurposed drugs have been screened and attempted against free parasites in clinical scenarios. Anti-leishmanial peptides are one such strategy that has recently gained in importance, particularly with active promotion strategies by pharmaceutical companies [31][32]. The commercialization of peptide-based drugs needs to rely heavily on their utility and clinical success with ease of synthesis, water solubility, sound biocompatibility, selectivity, versatility, tenability, and low toxicity [33]. Antimicrobial peptides (AMPs) are small molecules (<100 amino acids long) with positive charges and amphipathic specificities (hydrophobic and hydrophilic regions). AMPs act by affecting membranes by destabilization/disruption of phospholipids and induce cell death by increasing the permeability of the cell membrane and are less likely to be selective to resistant variants [34][35]. AMPs also pass through the membrane and interrupt or destabilize nucleic acid or protein synthesis and/or compromise enzyme (protease) functions or cell membrane synthesis. Thus, AMPs are an interesting candidate for effective therapeutic success against leishmaniasis [36]. The leishmanicidal effects of these peptides have been published in recent reports including the structural characteristics and inevitable challenges [37][38][39].
2. Chemotherapy in Leishmaniasis: Current Drugs, Limitations, and Challenges
The focus of this section relates to the discussion on the currently existing drugs in use for the treatment of VL. These include pentavalent antimonials, pentamidine, various formulations of amphotericin B (AmB), paromomycin, and miltefosine (Table 1 and Figure 1). These medications are also in use for the treatment of CL and MCL and PKDL. Treatment of VL considerably varies between the endemic regions spanning from India to Africa. The WHO approved and recommended regimens for known endemic VL foci are summarized in Table 1. Approximately 25 drugs or combinations are in use for humans with leishmaniasis [40][41][42].
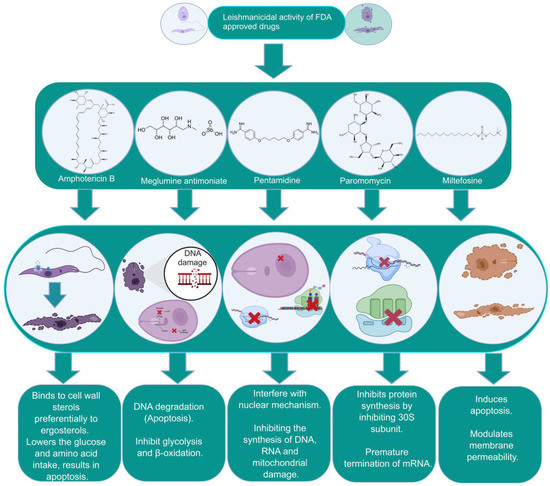
Figure 1. The biochemical characterization of FDA-approved drugs in Leishmania infection including the mode of action against the parasites.
WHO-OMS (2004) declared that liposomal amphotericin B, miltefosine, and paromomycin are the most promising drugs for the treatment of leishmanial infections. The search for potential new drugs and targets has been a very active area of research in the last couple of decades, with the publication of several important reviews [43][44][45][46][47][48][49][50][51][52][53][54][55][56].
Table 1. Details of the FDA-approved drugs for leishmaniasis, summarizing the cellular and molecular targets and limitations including side effects.
| Drugs | Structure | Comments | Efficacy | Resistance | Uses | Toxicity | Ref. |
|---|---|---|---|---|---|---|---|
| Meglumine antimoniate |
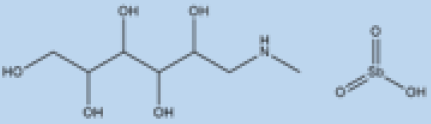 |
i.v. or i.m. First-line treatment. |
Varies between 35 and 95% based on area. |
High resistance in some regions of India. | VL, CL | Cardiotoxicity arthralgia, anorexia, fever, urticaria and significant toxicity to the liver, kidneys, and spleen. Hospitalization and constant monitoring of patients during treatment are needed. |
[57][58] |
| Paromomycin | 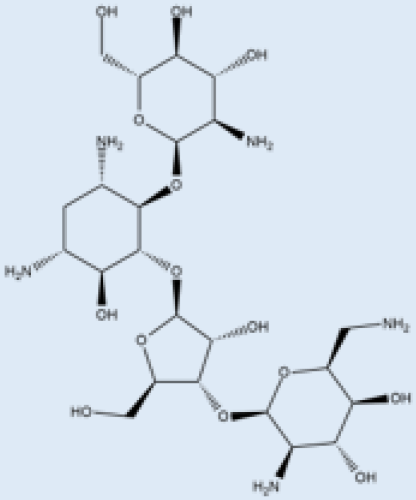 |
i.m. | A Phase III trial of Paromomycin (15 mg kg−1 (11 mg base) for 21 days showed 95% cure rate. Effective against PKDL. | No effective resistance. | CL, PKDL | Pain at the injection site, kidney toxicity, liver toxicity, and hearing toxicity. | [25][59] |
| Amphotericin B | 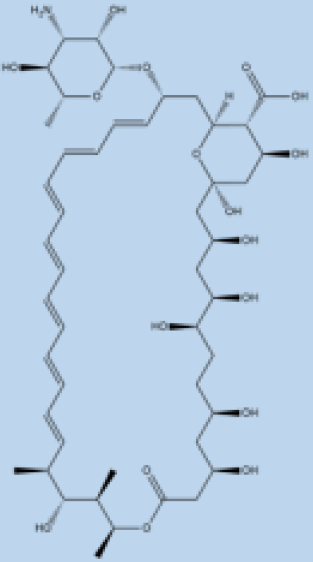 |
i.v. Very effective in regions with resistance. |
>90% | No effective resistance. | VL | Infusion-related reactions, anemia, nephrotoxicity, myocarditis, and even death of the patient. | [14][60] |
| Pentamidine |  |
Pentamidine is a second-line leishmaniasis treatment that is mostly used for CL. | With cure rates ranging from 35% with L. braziliensis in Peru to 90% with L. guyanensis in Suriname, efficacy is very variable. | Yes | CL, VL | Heart damage, joint pain, loss of appetite, fever, urticaria, and serious liver, kidneys, and spleen damage. During treatment, patients must be hospitalized and constantly watched. | [55][61] |
| Miltefosine |  |
p.o. Teratogenic. Increasing treatment failures. |
93–95% in India, 65–85% in Africa. | No effective resistance described. | CL, VL | It can cause birth defects, stomach problems, kidney damage, and liver damage and cannot be given to pregnant women. | [62][63] |
A wide range of compounds of multiple families have been identified as potential hits and leads, and some of which are in clinical trials. Several candidates such as inhibitors impairing thiol metabolism, sterol, glycolytic, folate and trypanothione metabolism, etc. are important to consider (Table 2). These drugs and the commonly used chemotherapy (Table 1) still lack the ability to provide efficient control against Leishmania. Several combinations have been employed in clinical practice [11][64] including less toxic drug delivery systems (DDSs) such as PLGA nanoparticles or liposomes [55], poly-aggregated forms of AmpB [65], or amphiphilic antimony [66]. Below, researchers describe in more detail the current treatment options including their inadequacies and the need for new chemical entities.
Table 2. Identification of novel antileishmanial drugs specific to biochemical pathways critical for the survival of Leishmania donovani.
| Pathway | Drug Target | Drug Candidate | Mode of Action |
Refs. |
|---|---|---|---|---|
| Sterol Biosynthesis Pathway | Squalene epoxidase | Spiro[indole-3,3′-pyrrolizidine]-2-one | DNA topoisomerase IB inhibitor. | [67][68] |
| HMGR enzyme | Mevastatin | Hampers HMGR activity. | [69][70] | |
| Sterol alpha-14 demethylase | Avodart | Induces ROS and causes apoptosis in the parasite. | [71] | |
| HMGR enzyme | Glycyrrhizic acid | Inhibits HMGR enzyme. | [72] | |
| Purine Salvage Pathway | mRNA translation | 5-fluorouracil 4-thiouracil |
Binds to RNA and blocks cell growth. | [73][74] |
| Glycolytic Pathway | GAPDH | Artesunate | Inhibits the parasites’ glycolytic enzymes GPDH. | [75][76] |
| Quinine | [75] | |||
| Mefloquine | [75] | |||
| Folate Biosynthesis Pathway | DHFR | Methotrexate (MTX, 1) | Inhibits DHFR. | [77] |
| Cycloguanil | [77] | |||
| Trimethoprim (TMP, 2) | [77][78] | |||
| ZINC57774418 (Z18) | Inhibits DHFR activity. | [79] | ||
| ZINC69844431 (Z31) | [79] | |||
| ZINC71746025 (Z25) | [79] | |||
| D11596 (DB96) | [79] | |||
| 3,4-dihydropyrimidine-2-one | [80] | |||
| 5-(3,5-dimethoxybenzyl) pyrimidine-2,4-diamine | [80] | |||
| DHFR and PTR1 | 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole | DHFR-TS/PTR1 inhibitors. | [81] | |
| 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole-1H-benzo[d]oxazole | [81] | |||
| Trypanothione Pathway | TR | Trichloro [1,2-ethanediolato-O,O’]-tellurate (AS101) | Induces ROS-mediated apoptosis by binding to TR cysteine residues. | [82] |
| β-sitosterol CCL | Inhibit TR activity. | [83] | ||
| Hypusine Pathway | Spermidine synthase | Hypericin | ROS and spermidine reduction. | [84][85] |
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970.
- Torpiano, P.; Pace, D. Leishmaniasis: Diagnostic Issues in Europe. Expert. Rev. Anti. Infect. Ther. 2015, 13, 1123–1138.
- Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 5 November 2022).
- Steverding, D. The History of Leishmaniasis. Parasites Vectors 2017, 10, 82.
- Melby, P.C.; Travi, B.L.; Yaneth Osorio, E. Leishmania. In Encyclopedia of Microbiology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 769–779. ISBN 978-0-12-811736-1.
- Gluenz, E.; Ginger, M.L.; McKean, P.G. Flagellum Assembly and Function during the Leishmania Life Cycle. Curr. Opin. Microbiol. 2010, 13, 473–479.
- Piscopo, T.V.; Mallia Azzopardi, C. Leishmaniasis. Postgrad. Med. J. 2007, 83, 649–657.
- Songumpai, N.; Promrangsee, C.; Noopetch, P.; Siriyasatien, P.; Preativatanyou, K. First Evidence of Co-Circulation of Emerging Leishmania Martiniquensis, Leishmania Orientalis, and Crithidia Sp. in Culicoides Biting Midges (Diptera: Ceratopogonidae), the Putative Vectors for Autochthonous Transmission in Southern Thailand. Trop. Med. Infect. Dis. 2022, 7, 379.
- Srivarasat, S.; Brownell, N.; Siriyasatien, P.; Noppakun, N.; Asawanonda, P.; Rattanakorn, K.; Preativatanyou, K.; Kumtornrut, C. Case Report: Autochthonous Disseminated Cutaneous, Mucocutaneous, and Visceral Leishmaniasis Caused by Leishmania Martiniquensis in a Patient with HIV/AIDS from Northern Thailand and Literature Review. Am. J. Trop. Med. Hyg. 2022, 107, 1196–1202.
- Alvar, J.; Arana, B.I. Appraisal of Leishmaniasis Chemotherapy, Current Status and Pipeline StrategiesChapter 1 Leishmaniasis, Impact and Therapeutic Needs; Royal Society of Chemistry: London, UK, 2017; pp. 1–23.
- Sangshetti, J.N.; Khan, F.A.K.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial Drug Discovery: Comprehensive Review of the Last 10 Years. RSC Adv. 2015, 5, 32376–32415.
- Lira, R.; Sundar, S.; Makharia, A.; Kenney, R.; Gam, A.; Saraiva, E.; Sacks, D. Evidence That the High Incidence of Treatment Failures in Indian Kala-Azar Is Due to the Emergence of Antimony-Resistant Strains of Leishmania Donovani. J. Infect. Dis. 1999, 180, 564–567.
- Sundar, S.; More, D.K.; Singh, M.K.; Singh, V.P.; Sharma, S.; Makharia, A.; Kumar, P.C.; Murray, H.W. Failure of Pentavalent Antimony in Visceral Leishmaniasis in India: Report from the Center of the Indian Epidemic. Clin. Infect. Dis. 2000, 31, 1104–1107.
- Sundar, S.; Chakravarty, J.; Agarwal, D.; Rai, M.; Murray, H.W. Single-Dose Liposomal Amphotericin B for Visceral Leishmaniasis in India. N. Engl. J. Med. 2010, 362, 504–512.
- Bern, C.; Adler-Moore, J.; Berenguer, J.; Boelaert, M.; den Boer, M.; Davidson, R.N.; Figueras, C.; Gradoni, L.; Kafetzis, D.A.; Ritmeijer, K.; et al. Liposomal Amphotericin B for the Treatment of Visceral Leishmaniasis. Clin. Infect. Dis. 2006, 43, 917–924.
- Meheus, F.; Balasegaram, M.; Olliaro, P.; Sundar, S.; Rijal, S.; Faiz, M.A.; Boelaert, M. Cost-Effectiveness Analysis of Combination Therapies for Visceral Leishmaniasis in the Indian Subcontinent. PLoS Negl. Trop. Dis. 2010, 4, e818.
- Croft, S.L.; Neal, R.A.; Pendergast, W.; Chan, J.H. The Activity of Alkyl Phosphorylcholines and Related Derivatives against Leishmania Donovani. Biochem. Pharmacol. 1987, 36, 2633–2636.
- Jha, T.K.; Sundar, S.; Thakur, C.P.; Bachmann, P.; Karbwang, J.; Fischer, C.; Voss, A.; Berman, J. Miltefosine, an Oral Agent, for the Treatment of Indian Visceral Leishmaniasis. N. Engl. J. Med. 1999, 341, 1795–1800.
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral Miltefosine for Indian Visceral Leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746.
- Rijal, S.; Ostyn, B.; Uranw, S.; Rai, K.; Bhattarai, N.R.; Dorlo, T.P.C.; Beijnen, J.H.; Vanaerschot, M.; Decuypere, S.; Dhakal, S.S.; et al. Increasing Failure of Miltefosine in the Treatment of Kala-Azar in Nepal and the Potential Role of Parasite Drug Resistance, Reinfection, or Noncompliance. Clin. Infect. Dis. 2013, 56, 1530–1538.
- Soto, J.; Toledo, J.; Gutierrez, P.; Nicholls, R.S.; Padilla, J.; Engel, J.; Fischer, C.; Voss, A.; Berman, J. Treatment of American Cutaneous Leishmaniasis with Miltefosine, an Oral Agent. Clin. Infect. Dis. 2001, 33, E57–E61.
- Soto, J.; Rea, J.; Balderrama, M.; Toledo, J.; Soto, P.; Valda, L.; Berman, J.D. Efficacy of Miltefosine for Bolivian Cutaneous Leishmaniasis. Am. J. Trop. Med. Hyg. 2008, 78, 210–211.
- Pinart, M.; Rueda, J.-R.; Romero, G.A.; Pinzón-Flórez, C.E.; Osorio-Arango, K.; Maia-Elkhoury, A.N.S.; Reveiz, L.; Elias, V.M.; Tweed, J.A. Interventions for American Cutaneous and Mucocutaneous Leishmaniasis. Cochrane Database Syst. Rev. 2020.
- Jha, T.K.; Olliaro, P.; Thakur, C.P.N.; Kanyok, T.P.; Singhania, B.L.; Singh, I.J.; Singh, N.K.P.; Akhoury, S.; Jha, S. Randomised Controlled Trial of Aminosidine (Paromomycin) v Sodium Stibogluconate for Treating Visceral Leishmaniasis in North Bihar, India. BMJ 1998, 316, 1200–1205.
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Sinha, P.K.; Bhattacharya, S.K. Injectable Paromomycin for Visceral Leishmaniasis in India. N. Engl. J. Med. 2007, 356, 2571–2581.
- Hailu, A.; Musa, A.; Wasunna, M.; Balasegaram, M.; Yifru, S.; Mengistu, G.; Hurissa, Z.; Hailu, W.; Weldegebreal, T.; Tesfaye, S.; et al. Geographical Variation in the Response of Visceral Leishmaniasis to Paromomycin in East Africa: A Multicentre, Open-Label, Randomized Trial. PLoS Negl. Trop. Dis. 2010, 4, e709.
- Musa, A.M.; Younis, B.; Fadlalla, A.; Royce, C.; Balasegaram, M.; Wasunna, M.; Hailu, A.; Edwards, T.; Omollo, R.; Mudawi, M.; et al. Paromomycin for the Treatment of Visceral Leishmaniasis in Sudan: A Randomized, Open-Label, Dose-Finding Study. PLoS Negl. Trop. Dis. 2010, 4, e855.
- Fernández, O.L.; Diaz-Toro, Y.; Ovalle, C.; Valderrama, L.; Muvdi, S.; Rodríguez, I.; Gomez, M.A.; Saravia, N.G. Miltefosine and Antimonial Drug Susceptibility of Leishmania Viannia Species and Populations in Regions of High Transmission in Colombia. PLoS Negl. Trop. Dis. 2014, 8, e2871.
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful Decade for Antileishmanial Compounds from 2002 to Late 2011. Chem. Rev. 2014, 114, 10369–10428.
- Sundar, S.; Chakravarty, J.; Meena, L.P. Leishmaniasis: Treatment, Drug Resistance and Emerging Therapies. Expert. Opin. Orphan Drugs 2019, 7, 1–10.
- Zahedifard, F.; Rafati, S. Prospects for Antimicrobial Peptide-Based Immunotherapy Approaches in Leishmania Control. Expert. Rev. Anti. Infect. Ther. 2018, 16, 461–469.
- Rafferty, J.; Nagaraj, H.; McCloskey, A.P.; Huwaitat, R.; Porter, S.; Albadr, A.; Laverty, G. Peptide Therapeutics and the Pharmaceutical Industry: Barriers Encountered Translating from the Laboratory to Patients. Curr. Med. Chem. 2016, 23, 4231–4259.
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the Use of Therapeutic Peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21.
- Costa, F.; Teixeira, C.; Gomes, P.; Martins, M.C.L. Clinical Application of AMPs. Adv. Exp. Med. Biol. 2019, 1117, 281–298.
- Almeida, J.R.; Mendes, B.; Lancellotti, M.; Marangoni, S.; Vale, N.; Passos, Ó.; Ramos, M.J.; Fernandes, P.A.; Gomes, P.; Da Silva, S.L. A Novel Synthetic Peptide Inspired on Lys49 Phospholipase A2 from Crotalus Oreganus Abyssus Snake Venom Active against Multidrug-Resistant Clinical Isolates. Eur. J. Med. Chem. 2018, 149, 248–256.
- Cobb, S.L.; Denny, P.W. Antimicrobial Peptides for Leishmaniasis. Curr. Opin. Investig. Drugs. 2010, 11, 868–875.
- Torrent, M.; Pulido, D.; Rivas, L.; Andreu, D. Antimicrobial Peptide Action on Parasites. Curr. Drug. Targets 2012, 13, 1138–1147.
- Marr, A.K.; Cen, S.; Hancock, R.E.W.; McMaster, W.R. Identification of Synthetic and Natural Host Defense Peptides with Leishmanicidal Activity. Antimicrob. Agents Chemother. 2016, 60, 2484–2491.
- Luque-Ortega, J.R.; Rivas, L. Characterization of the Leishmanicidal Activity of Antimicrobial Peptides. Methods Mol. Biol. 2010, 618, 393–420.
- González, U.; Pinart, M.; Reveiz, L.; Alvar, J. Interventions for Old World Cutaneous Leishmaniasis. Cochrane Database Syst. Rev. 2008, CD005067.
- Sundar, S.; Singh, A. Chemotherapeutics of Visceral Leishmaniasis: Present and Future Developments. Parasitology 2018, 145, 481–489.
- Passero, L.F.D.; Brunelli, E.D.S.; Sauini, T.; Amorim Pavani, T.F.; Jesus, J.A.; Rodrigues, E. The Potential of Traditional Knowledge to Develop Effective Medicines for the Treatment of Leishmaniasis. Front. Pharmacol. 2021, 12, 690432.
- Monzote, L. Current Treatment of Leishmaniasis: A Review. Open. Antimicrob. Agents J. 2009, 1, 9–19.
- Freitas-Junior, L.H.; Chatelain, E.; Kim, H.A.; Siqueira-Neto, J.L. Visceral Leishmaniasis Treatment: What Do We Have, What Do We Need and How to Deliver It? Int. J. Parasitol. Drugs Drug. Resist. 2012, 2, 11–19.
- No, J.H. Visceral Leishmaniasis: Revisiting Current Treatments and Approaches for Future Discoveries. Acta Trop. 2016, 155, 113–123.
- Singh, K.; Garg, G.; Ali, V. Current Therapeutics, Their Problems and Thiol Metabolism as Potential Drug Targets in Leishmaniasis. Curr. Drug. Metab. 2016, 17, 897–919.
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug Resistance and Treatment Failure in Leishmaniasis: A 21st Century Challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052.
- Rivas, L.; Gil, G. Drug Discovery for Leishmaniasis; Royal Society of Chemistry: London, UK, 2018.
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Muthoni Ouattara, G.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent Development of Visceral Leishmaniasis Treatments: Successes, Pitfalls, and Perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18.
- Andrade-Neto, V.V.; Cunha-Junior, E.F.; Dos Santos Faioes, V.; Pereira, T.M.; Silva, R.L.; Leon, L.L.; Torres-Santos, E.C. Leishmaniasis Treatment: Update of Possibilities for Drug Repurposing. Front. Biosci. Landmark 2018, 23, 967–996.
- Reguera, R.M.; Pérez-Pertejo, Y.; Gutiérrez-Corbo, C.; Domínguez-Asenjo, B.; Ordóñez, C.; García-Estrada, C.; Martínez-Valladares, M.; Balaña-Fouce, R. Current and Promising Novel Drug Candidates against Visceral Leishmaniasis. Pure Appl. Chem. 2019, 91, 1385–1404.
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of Leishmaniasis: Present Challenges. Parasitology 2018, 145, 464–480.
- Sangenito, L.S.; da Silva Santos, V.; d’Avila-Levy, C.M.; Branquinha, M.H.; Souza Dos Santos, A.L.; de Oliveira, S.S.C. Leishmaniasis and Chagas Disease-Neglected Tropical Diseases: Treatment Updates. Curr. Top. Med. Chem. 2019, 19, 174–177.
- van Griensven, J.; Diro, E. Visceral Leishmaniasis: Recent Advances in Diagnostics and Treatment Regimens. Infect. Dis. Clin. N. Am. 2019, 33, 79–99.
- J, B.; M, B.M.; Chanda, K. An Overview on the Therapeutics of Neglected Infectious Diseases—Leishmaniasis and Chagas Diseases. Front. Chem. 2021, 9, 622286.
- Sasidharan, S.; Saudagar, P. Leishmaniasis: Where Are We and Where Are We Heading? Parasitol. Res. 2021, 120, 1541–1554.
- Sundar, S.; Chakravarty, J.; Rai, V.K.; Agrawal, N.; Singh, S.P.; Chauhan, V.; Murray, H.W. Amphotericin B Treatment for Indian Visceral Leishmaniasis: Response to 15 Daily versus Alternate-Day Infusions. Clin. Infect. Dis. 2007, 45, 556–561.
- de Oliveira, A.L.L.; Brustoloni, Y.M.; Fernandes, T.D.; Dorval, M.E.C.; da Cunha, R.V.; Bóia, M.N. Severe Adverse Reactions to Meglumine Antimoniate in the Treatment of Visceral Leishmaniasis: A Report of 13 Cases in the Southwestern Region of Brazil. Trop. Doct. 2009, 39, 180–182.
- Sundar, S.; Singh, A.; Tiwari, A.; Shukla, S.; Chakravarty, J.; Rai, M. Efficacy and Safety of Paromomycin in Treatment of Post-Kala-Azar Dermal Leishmaniasis. ISRN Parasitol. 2014, 2014, 548010.
- de Menezes, J.P.B.; Guedes, C.E.S.; de Oliveira Almeida Petersen, A.L.; Fraga, D.B.M.; Veras, P.S.T. Advances in Development of New Treatment for Leishmaniasis. BioMed Res. Int. 2015, 2015, e815023.
- Andersen, E.M.; Cruz-Saldarriaga, M.; Llanos-Cuentas, A.; Luz-Cjuno, M.; Echevarria, J.; Miranda-Verastegui, C.; Colina, O.; Berman, J.D. Comparison of Meglumine Antimoniate and Pentamidine for Peruvian Cutaneous Leishmaniasis. Am. J. Trop. Med. Hyg. 2005, 72, 133–137.
- Bhattacharya, S.K.; Sinha, P.K.; Sundar, S.; Thakur, C.P.; Jha, T.K.; Pandey, K.; Das, V.R.; Kumar, N.; Lal, C.; Verma, N.; et al. Phase 4 Trial of Miltefosine for the Treatment of Indian Visceral Leishmaniasis. J. Infect. Dis. 2007, 196, 591–598.
- Antinori, S.; Schifanella, L.; Corbellino, M. Leishmaniasis: New Insights from an Old and Neglected Disease. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 109–118.
- Le Pape, P. Development of New Antileishmanial Drugs--Current Knowledge and Future Prospects. J. Enzyme Inhib. Med. Chem. 2008, 23, 708–718.
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B Formulations and Drug Targeting. J. Pharm. Sci. 2008, 97, 2405–2425.
- Fernandes, F.R.; Ferreira, W.A.; Campos, M.A.; Ramos, G.S.; Kato, K.C.; Almeida, G.G.; Corrêa, J.D.; Melo, M.N.; Demicheli, C.; Frézard, F. Amphiphilic Antimony(V) Complexes for Oral Treatment of Visceral Leishmaniasis. Antimicrob. Agents Chemother. 2013, 57, 4229–4236.
- Saha, S.; Acharya, C.; Pal, U.; Chowdhury, S.R.; Sarkar, K.; Maiti, N.C.; Jaisankar, P.; Majumder, H.K. A Novel Spirooxindole Derivative Inhibits the Growth of Leishmania Donovani Parasites Both In Vitro and In Vivo by Targeting Type IB Topoisomerase. Antimicrob. Agents Chemother. 2016, 60, 6281–6293.
- Leañez, J.; Nuñez, J.; García-Marchan, Y.; Sojo, F.; Arvelo, F.; Rodriguez, D.; Buscema, I.; Alvarez-Aular, A.; Bello Forero, J.S.; Kouznetsov, V.V.; et al. Anti-Leishmanial Effect of Spiro Dihydroquinoline-Oxindoles on Volume Regulation Decrease and Sterol Biosynthesis of Leishmania Braziliensis. Exp. Parasitol. 2019, 198, 31–38.
- Dinesh, N.; Soumya, N.; Singh, S. Antileishmanial Effect of Mevastatin Is Due to Interference with Sterol Metabolism. Parasitol. Res. 2015, 114, 3873–3883.
- Singh, S.; Babu, N.K.; Singh, S.; Babu, N.K. 3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGR) Enzyme of the Sterol Biosynthetic Pathway: A Potential Target against Visceral Leishmaniasis; IntechOpen: London, UK, 2018; ISBN 978-1-78984-102-2.
- Tabrez, S.; Rahman, F.; Ali, R.; Akand, S.K.; Alaidarous, M.A.; Alshehri, B.M.; Banawas, S.; Dukhyil, A.A.B.; Rub, A. Targeting Sterol Alpha-14 Demethylase of Leishmania Donovani to Fight against Leishmaniasis. J. Cell Biochem. 2021, 122, 1037–1047.
- Dinesh, N.; Neelagiri, S.; Kumar, V.; Singh, S. Glycyrrhizic Acid Attenuates Growth of Leishmania Donovani by Depleting Ergosterol Levels. Exp. Parasitol. 2017, 176, 21–29.
- Azzouz, S.; Lawton, P. In Vitro Effects of Purine and Pyrimidine Analogues on Leishmania Donovani and Leishmania Infantum Promastigotes and Intracellular Amastigotes. Acta Parasitol. 2017, 62, 582–588.
- Soysa, R.; Wilson, Z.N.; Elferich, J.; Forquer, I.; Shinde, U.; Riscoe, M.K.; Yates, P.A.; Ullman, B. Substrate Inhibition of Uracil Phosphoribosyltransferase by Uracil Can Account for the Uracil Growth Sensitivity of Leishmania Donovani Pyrimidine Auxotrophs. J. Biol. Chem. 2013, 288, 29954–29964.
- Yousef, B.; Elwaseela, T.; Ali, T.; Mohammed, F.; Mohammed, W.; Alobaid, M.; Ibrahim Dirar, A. Anti-Malarial Drugs as Potential Inhibitors of Leishmania Glycolytic Enzymes: Development of New Anti-Leishmanial Agents. Pharmacol. Clin. Pharm. Res. 2020, 5, 77–88.
- Verma, A.; Ghosh, S.; Salotra, P.; Singh, R. Artemisinin-Resistant Leishmania Parasite Modulates Host Cell Defense Mechanism and Exhibits Altered Expression of Unfolded Protein Response Genes. Parasitol. Res. 2019, 118, 2705–2713.
- Hendrickx, S.; Caljon, G.; Maes, L. Need for Sustainable Approaches in Antileishmanial Drug Discovery. Parasitol. Res. 2019, 118, 2743–2752.
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and Other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J. Antibiot. 2020, 73, 5–27.
- Sharma, V.K.; Bharatam, P.V. Identification of Selective Inhibitors of LdDHFR Enzyme Using Pharmacoinformatic Methods. J. Comput. Biol. 2021, 28, 43–59.
- das Neves, G.M.; Kagami, L.P.; Gonçalves, I.L.; Eifler-Lima, V.L. Targeting Pteridine Reductase 1 and Dihydrofolate Reductase: The Old Is a New Trend for Leishmaniasis Drug Discovery. Future Med. Chem. 2019, 11, 2107–2130.
- Kapil, S.; Singh, P.K.; Kashyap, A.; Silakari, O. Structure Based Designing of Benzimidazole/Benzoxazole Derivatives as Anti-Leishmanial Agents. SAR QSAR Environ. Res. 2019, 30, 919–933.
- Vishwakarma, P.; Parmar, N.; Chandrakar, P.; Sharma, T.; Kathuria, M.; Agnihotri, P.K.; Siddiqi, M.I.; Mitra, K.; Kar, S. Ammonium Trichloro -Tellurate Cures Experimental Visceral Leishmaniasis by Redox Modulation of Leishmania Donovani Trypanothione Reductase and Inhibiting Host Integrin Linked PI3K/Akt Pathway. Cell. Mol. Life Sci. 2018, 75, 563–588.
- Pramanik, P.K.; Chakraborti, S.; Bagchi, A.; Chakraborti, T. Bioassay-Based Corchorus Capsularis L. Leaf-Derived β-Sitosterol Exerts Antileishmanial Effects against Leishmania Donovani by Targeting Trypanothione Reductase. Sci. Rep. 2020, 10, 20440.
- Singh, S.; Kumari, E.; Bhardwaj, R.; Kumar, R.; Dubey, V.K. Molecular Events Leading to Death of Leishmania Donovani under Spermidine Starvation after Hypericin Treatment. Chem. Biol. Drug. Des. 2017, 90, 962–971.
- Singh, S.; Sarma, S.; Katiyar, S.P.; Das, M.; Bhardwaj, R.; Sundar, D.; Dubey, V.K. Probing the Molecular Mechanism of Hypericin-Induced Parasite Death Provides Insight into the Role of Spermidine beyond Redox Metabolism in Leishmania Donovani. Antimicrob. Agents Chemother. 2015, 59, 15–24.
More
Information
Subjects:
Zoology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
738
Revisions:
2 times
(View History)
Update Date:
24 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No