Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Zoology
Leishmaniasis, a category 1 neglected protozoan disease caused by a kinetoplastid pathogen called Leishmania, is transmitted through dipteran insect vectors (phlebotomine, sand flies) in three main clinical forms: fatal visceral leishmaniasis, self-healing cutaneous leishmaniasis, and mucocutaneous leishmaniasis.
- chemoinformatics
- bioinformatics
- drugs
- inhibitors
1. Introduction
Leishmaniasis is a group of vector-borne infectious protozoan diseases endemic to nearly one hundred countries [1]. Leishmaniasis is considered by the WHO to be a neglected tropical disease and a major international health challenge. In addition to malaria, it is the deadliest parasitic disease worldwide. Nearly 0.71–1 million new cases and approximately 20,000 to 65,000 deaths are reported annually, predominantly in socioeconomically vulnerable communities with limited access to essential medicines. Over 20 different species of Leishmania are reported to be infective to humans, categorized as Old World (Mediterranean countries, Asia, and Africa) and New World (America) forms. The dipteran fly Phlebotomus and its subspecies in the Old World and Lutzomyia in the New World are proven vectors for human leishmaniasis. The disease globally affects approximately 14 million people, with over one billion people at high risk of infection [2]. A World Health Organization (WHO)-sponsored epidemiological report indicates that there are nearly 12 million active cases of leishmaniasis. The incidence of cutaneous leishmaniasis is two to three times more common than visceral leishmaniasis [3,4]. At present, this disease results in 20,000 to 65,000 deaths reported annually and is included among the 18 most neglected tropical diseases (NTDs). Approximately one hundred species of these dipteran insects belonging to the genera Phlebotomus and Lutzomyia are known as the main vectors involved in biological transmission [4]. Leishmania has a complex life cycle characterized by the presence of digenetic stages: flagellated promastigotes and flagellated amastigotes [5]. The metacyclic promastigote form in sand flies is responsible for infection in healthy individuals. The amastigote form is known for its pathogenesis, having a spherical shape with a rudimentary flagellum. Amastigotes reside, propagate, and persist within the host’s mononuclear phagocytic cells [6]. Leishmaniasis represents a wide spectrum pathology ranging from less severe and self-curable cutaneous leishmaniasis (CL) to more severe and fatal visceral leishmaniasis (VL). The clinical symptoms due to parasitic infections are classified into three types of disease: cutaneous, mucocutaneous, and visceral leishmaniasis [5,6]. Cutaneous leishmaniasis is manifested by the development of skin lesions and is the most common type prevalent in the Middle East. Visceral leishmaniasis, on the other hand, is distinguished by the occurrence of hepatosplenomegaly, fever, and weight loss, and is considered as a serious health hazard for the infected individual. Mucocutaneous leishmaniasis (MCL) is characterized by damage to oral mucous membranes in the nose, mouth, and throat, which potentiates inflammation and face disfiguration [7]. Recently, a new subgenus, Mundinia, has been reported, and a member of this group (L. martiniquensis) causes VL in Southeast Asian regions. L. martiniquensis typically causes VL in humans and can be treated with amphotericin B as a first-line chemotherapeutic option. It has been reported that VL caused by L. martiniquensis has a higher relapse rate and occurs in individuals with HIV infection [8,9].
In addition to two other kinetoplastid pathogens, viz. for Trypanosoma cruzi and Trypanosoma brucei, the management of leishmaniasis requires integrated and multidisciplinary strategies that include vector control, enhanced diagnostics, and increased awareness of new therapies with safe and efficient medicines [10]. There is still no effective vaccine available, and the control of the disease primarily rests on chemotherapy, the majority of which is costly and has a wide array of side effects [10,11]. Pentavalent antimonials (sodium stibogluconate, meglumine antimoniate or generic formulations) have been used as standard drugs in countries such as India and Nepal for over 60 years and remain the primary treatment options in many endemic regions despite widespread parasite resistance [12,13]. A single dose of the polyene antibiotic amphotericin B demonstrated a 95% efficiency against visceral leishmaniasis in India [14]. Intravenous administration of liposomal amphotericin B has become a standard treatment in many countries but remains expensive, even for single-course treatments [15,16]. Miltefosine, an alkyl-lysophospholipid analog, was initially developed as an anticancer compound and is considered as a first-line effective oral drug against Leishmania [17,18]. Miltefosine has been used successfully for the treatment of VL in India since 2002 and has been incorporated into the visceral leishmaniasis elimination program for the Indian subcontinent [19]. Despite some success, miltefosine administration registers considerable resistance, with relapse in nearly 20% of patients post-treatment [20]. Miltefosine was also found to be effective against cutaneous and mucocutaneous leishmaniasis in South America, with considerable differences in percent success in therapy programs [21,22,23]. Recently, the aminoglycoside paromomycin has been approved for the treatment of VL. The efficacy of paromomycin against VL patients has been demonstrated in phase III clinical trials in India [24,25]. However, paromomycin has also shown shortcomings in efficacy across geographical regions, as demonstrated by its less than satisfactory trial in Sudan compared to India [26,27]. It is known that the drugs used act via different cellular and molecular mechanisms, causing a variety of outcomes including the apoptosis of parasites, but are also widely associated with variable toxicity and setbacks in sought-after results. These difficulties are further complicated by the emergence of drug resistance against parasites across the globe that have persisted alongside conventional chemotherapy practiced in endemic areas. This significantly lowers the susceptibility to drugs and the emergence of difficult-to-treat resistant variants of the same species [28,29,30].
Many investigators have highlighted the need to discover new drug targets employing the knowledge of parasite biochemistry to develop revolutionary new drugs by using emerging technologies. Several natural and synthetic drugs as well as repurposed drugs have been screened and attempted against free parasites in clinical scenarios. Anti-leishmanial peptides are one such strategy that has recently gained in importance, particularly with active promotion strategies by pharmaceutical companies [31,32]. The commercialization of peptide-based drugs needs to rely heavily on their utility and clinical success with ease of synthesis, water solubility, sound biocompatibility, selectivity, versatility, tenability, and low toxicity [33]. Antimicrobial peptides (AMPs) are small molecules (<100 amino acids long) with positive charges and amphipathic specificities (hydrophobic and hydrophilic regions). AMPs act by affecting membranes by destabilization/disruption of phospholipids and induce cell death by increasing the permeability of the cell membrane and are less likely to be selective to resistant variants [34,35]. AMPs also pass through the membrane and interrupt or destabilize nucleic acid or protein synthesis and/or compromise enzyme (protease) functions or cell membrane synthesis. Thus, AMPs are an interesting candidate for effective therapeutic success against leishmaniasis [36]. The leishmanicidal effects of these peptides have been published in recent reports including the structural characteristics and inevitable challenges [37,38,39].
2. Chemotherapy in Leishmaniasis: Current Drugs, Limitations, and Challenges
The focus of this section relates to the discussion on the currently existing drugs in use for the treatment of VL. These include pentavalent antimonials, pentamidine, various formulations of amphotericin B (AmB), paromomycin, and miltefosine (Table 1 and Figure 1). These medications are also in use for the treatment of CL and MCL and PKDL. Treatment of VL considerably varies between the endemic regions spanning from India to Africa. The WHO approved and recommended regimens for known endemic VL foci are summarized in Table 1. Approximately 25 drugs or combinations are in use for humans with leishmaniasis [40,41,42].
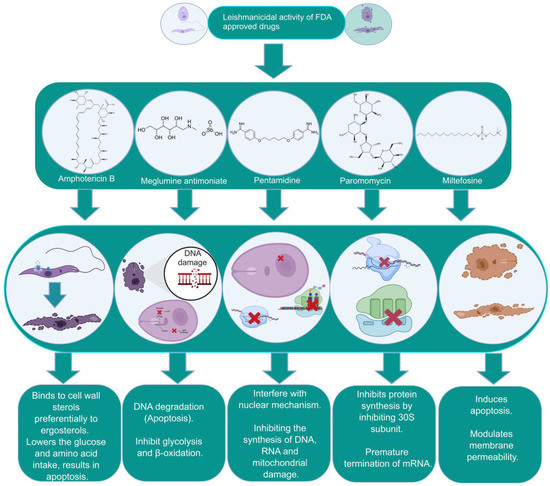
Figure 1. The biochemical characterization of FDA-approved drugs in Leishmania infection including the mode of action against the parasites.
WHO-OMS (2004) declared that liposomal amphotericin B, miltefosine, and paromomycin are the most promising drugs for the treatment of leishmanial infections. The search for potential new drugs and targets has been a very active area of research in the last couple of decades, with the publication of several important reviews [43,44,45,46,47,48,49,50,51,52,53,54,55,56].
Table 1. Details of the FDA-approved drugs for leishmaniasis, summarizing the cellular and molecular targets and limitations including side effects.
| Drugs | Structure | Comments | Efficacy | Resistance | Uses | Toxicity | Ref. |
|---|---|---|---|---|---|---|---|
| Meglumine antimoniate |
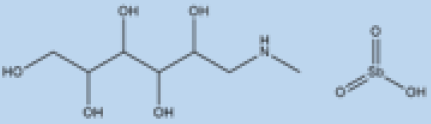 |
i.v. or i.m. First-line treatment. |
Varies between 35 and 95% based on area. |
High resistance in some regions of India. | VL, CL | Cardiotoxicity arthralgia, anorexia, fever, urticaria and significant toxicity to the liver, kidneys, and spleen. Hospitalization and constant monitoring of patients during treatment are needed. |
[57,58] |
| Paromomycin | 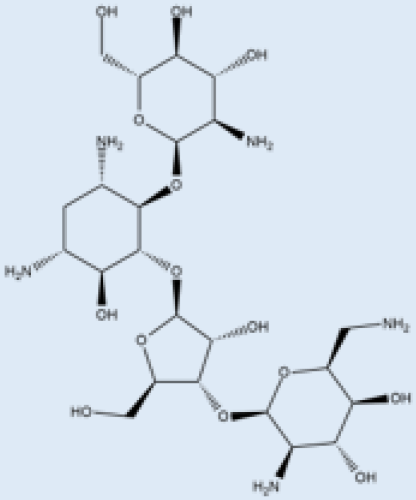 |
i.m. | A Phase III trial of Paromomycin (15 mg kg−1 (11 mg base) for 21 days showed 95% cure rate. Effective against PKDL. | No effective resistance. | CL, PKDL | Pain at the injection site, kidney toxicity, liver toxicity, and hearing toxicity. | [25,59] |
| Amphotericin B | 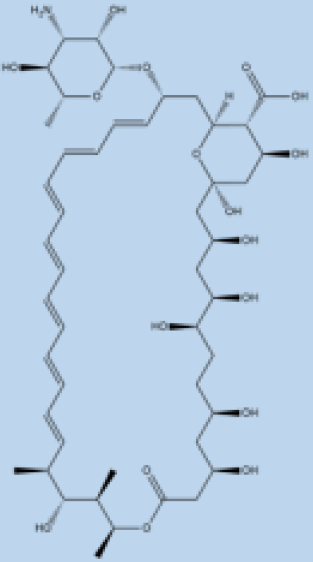 |
i.v. Very effective in regions with resistance. |
>90% | No effective resistance. | VL | Infusion-related reactions, anemia, nephrotoxicity, myocarditis, and even death of the patient. | [14,60] |
| Pentamidine |  |
Pentamidine is a second-line leishmaniasis treatment that is mostly used for CL. | With cure rates ranging from 35% with L. braziliensis in Peru to 90% with L. guyanensis in Suriname, efficacy is very variable. | Yes | CL, VL | Heart damage, joint pain, loss of appetite, fever, urticaria, and serious liver, kidneys, and spleen damage. During treatment, patients must be hospitalized and constantly watched. | [55,61] |
| Miltefosine |  |
p.o. Teratogenic. Increasing treatment failures. |
93–95% in India, 65–85% in Africa. | No effective resistance described. | CL, VL | It can cause birth defects, stomach problems, kidney damage, and liver damage and cannot be given to pregnant women. | [62,63] |
A wide range of compounds of multiple families have been identified as potential hits and leads, and some of which are in clinical trials. Several candidates such as inhibitors impairing thiol metabolism, sterol, glycolytic, folate and trypanothione metabolism, etc. are important to consider (Table 2). These drugs and the commonly used chemotherapy (Table 1) still lack the ability to provide efficient control against Leishmania. Several combinations have been employed in clinical practice [11,64] including less toxic drug delivery systems (DDSs) such as PLGA nanoparticles or liposomes [55], poly-aggregated forms of AmpB [65], or amphiphilic antimony [66]. Below, we describe in more detail the current treatment options including their inadequacies and the need for new chemical entities.
Table 2. Identification of novel antileishmanial drugs specific to biochemical pathways critical for the survival of Leishmania donovani.
| Pathway | Drug Target | Drug Candidate | Mode of Action |
Refs. |
|---|---|---|---|---|
| Sterol Biosynthesis Pathway | Squalene epoxidase | Spiro[indole-3,3′-pyrrolizidine]-2-one | DNA topoisomerase IB inhibitor. | [67,68] |
| HMGR enzyme | Mevastatin | Hampers HMGR activity. | [69,70] | |
| Sterol alpha-14 demethylase | Avodart | Induces ROS and causes apoptosis in the parasite. | [71] | |
| HMGR enzyme | Glycyrrhizic acid | Inhibits HMGR enzyme. | [72] | |
| Purine Salvage Pathway | mRNA translation | 5-fluorouracil 4-thiouracil |
Binds to RNA and blocks cell growth. | [73,74] |
| Glycolytic Pathway | GAPDH | Artesunate | Inhibits the parasites’ glycolytic enzymes GPDH. | [75,76] |
| Quinine | [75] | |||
| Mefloquine | [75] | |||
| Folate Biosynthesis Pathway | DHFR | Methotrexate (MTX, 1) | Inhibits DHFR. | [77] |
| Cycloguanil | [77] | |||
| Trimethoprim (TMP, 2) | [77,78] | |||
| ZINC57774418 (Z18) | Inhibits DHFR activity. | [79] | ||
| ZINC69844431 (Z31) | [79] | |||
| ZINC71746025 (Z25) | [79] | |||
| D11596 (DB96) | [79] | |||
| 3,4-dihydropyrimidine-2-one | [80] | |||
| 5-(3,5-dimethoxybenzyl) pyrimidine-2,4-diamine | [80] | |||
| DHFR and PTR1 | 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole | DHFR-TS/PTR1 inhibitors. | [81] | |
| 2-(4-((2,4-dichlorobenzyl)oxy)phenyl)-1H-benzo[d]imidazole-1H-benzo[d]oxazole | [81] | |||
| Trypanothione Pathway | TR | Trichloro [1,2-ethanediolato-O,O’]-tellurate (AS101) | Induces ROS-mediated apoptosis by binding to TR cysteine residues. | [82] |
| β-sitosterol CCL | Inhibit TR activity. | [83] | ||
| Hypusine Pathway | Spermidine synthase | Hypericin | ROS and spermidine reduction. | [84,85] |
This entry is adapted from the peer-reviewed paper 10.3390/pathogens12050706
This entry is offline, you can click here to edit this entry!