 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Puneetpal Singh | -- | 2617 | 2023-04-24 06:12:48 | | | |
| 2 | Catherine Yang | Meta information modification | 2617 | 2023-04-24 07:16:29 | | |
Video Upload Options
Atherosclerosis is the formation of plaque within arteries due to overt assemblage of fats, cholesterol and fibrous material causing a blockage of the free flow of blood leading to ischemia. It is harshly impinging on health statistics worldwide because of being principal cause of high morbidity and mortality for several diseases including rheumatological, heart and brain disorders. Atherosclerosis is perpetuated by pro-inflammatory and exacerbated by pro-coagulatory mediators. Besides several other pathways, the formation of neutrophil extracellular traps (NETs) and the activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome contribute significantly to the initiation and propagation of atherosclerotic plaque for its worst outcomes.
1. Introduction
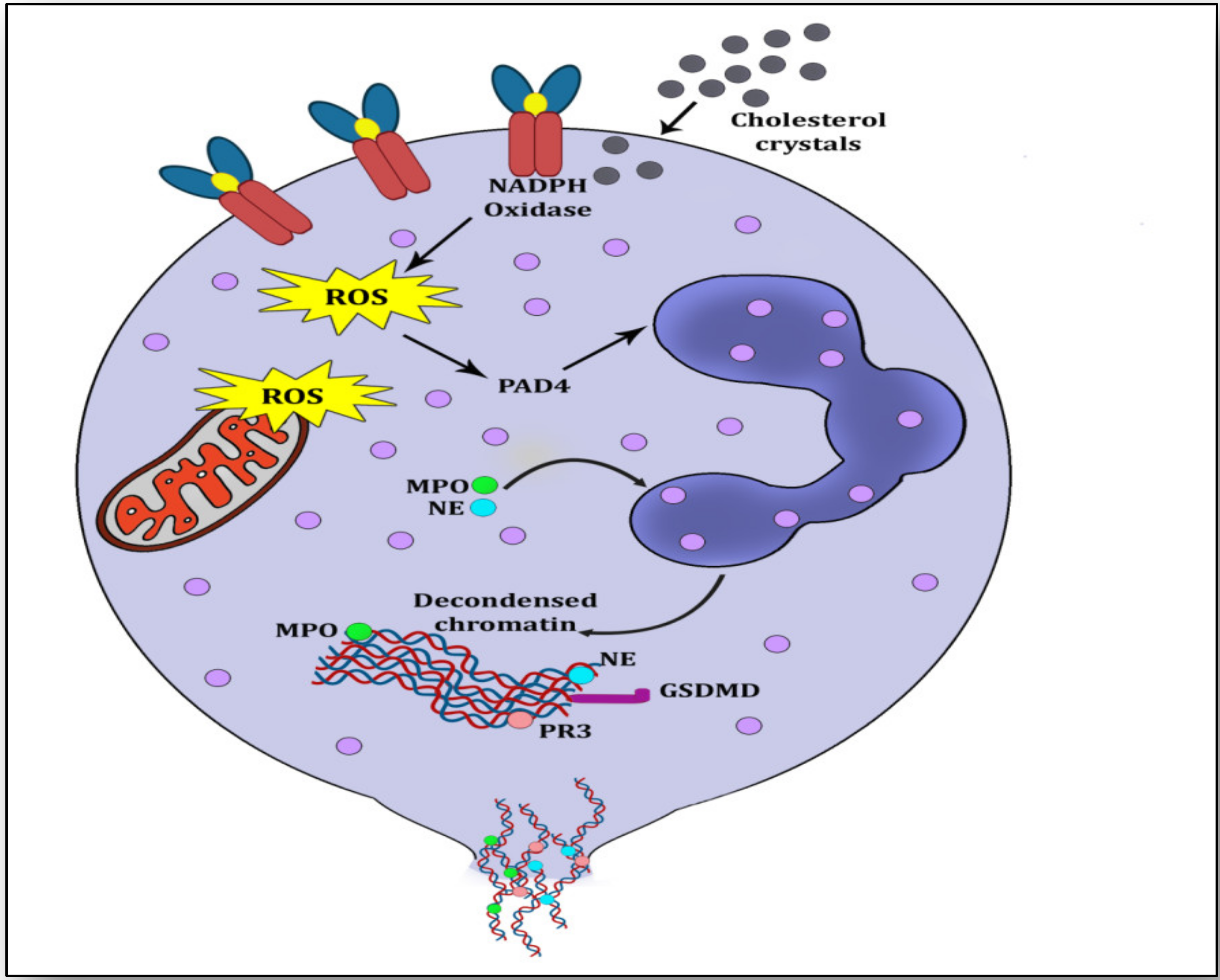
2. NETs, NETosis and Atherosclerosis
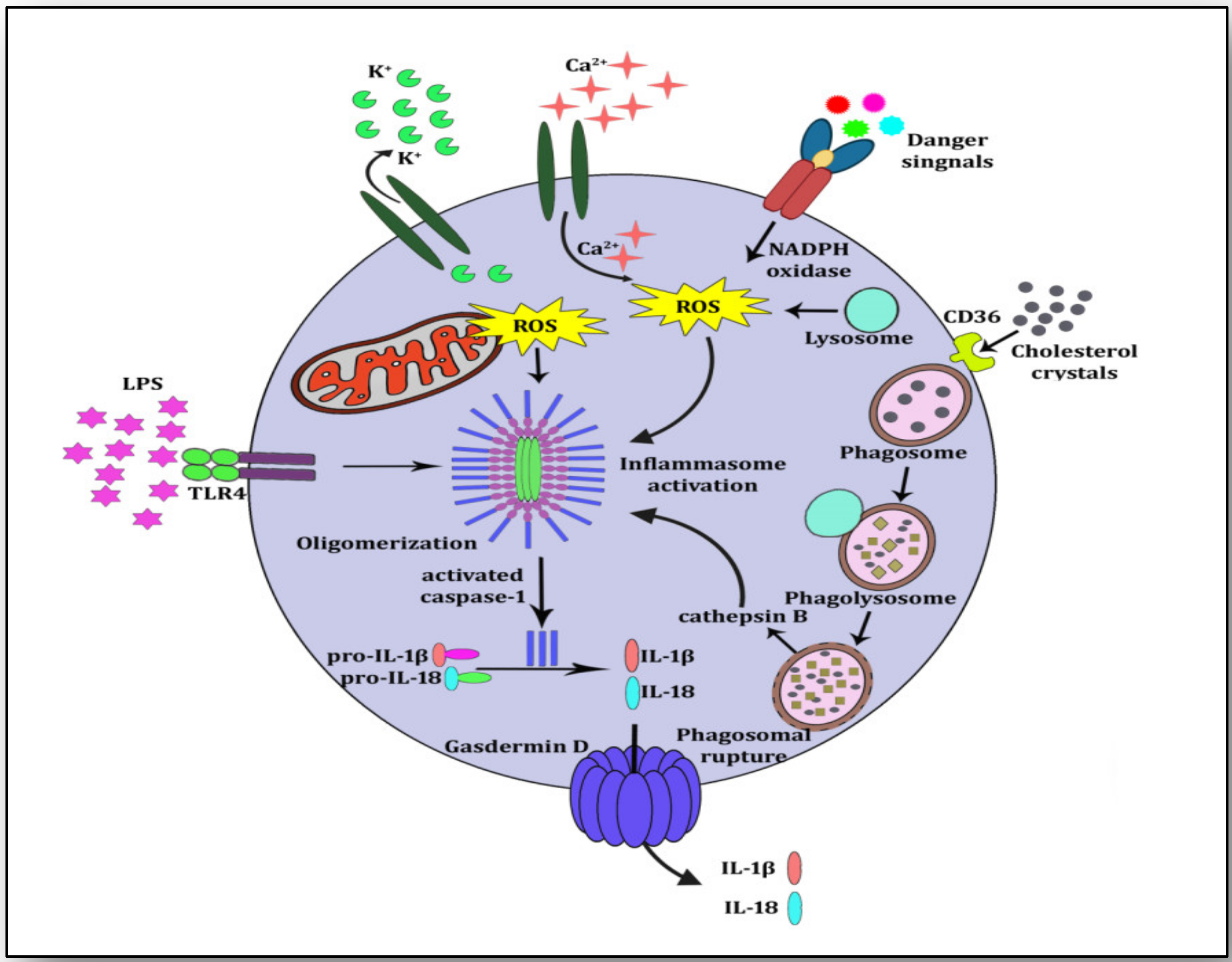
3. NLRP3 Inflammasome Activation and Atherosclerosis
4. NETosis-NLRP3 Inflammasome Activation Link: A Maleficent Crosstalk

References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56.
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406.
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell. Biol. 2015, 209, 13–22.
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131.
- Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231.
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PLoS ONE 2012, 7, e48111.
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287.
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743.
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl. Acad. Sci. USA 2020, 117, 7326–7337.
- Damascena, H.L.; Silveira, W.A.A.; Castro, M.S.; Fontes, W. Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate). Cells 2022, 11, 2889.
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025.
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469.
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell. Biol. 2010, 191, 677–691.
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32.
- Dabagh, M.; Jalali, P.; Tarbell, J.M. The transport of LDL across the deformable arterial wall: The effect of endothelial cell turnover and intimal deformation under hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H983–H996.
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956.
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598.
- Chrysanthopoulou, A.; Gkaliagkousi, E.; Lazaridis, A.; Arelaki, S.; Pateinakis, P.; Ntinopoulou, M.; Mitsios, A.; Antoniadou, C.; Argyriou, C.; Georgiadis, G.S.; et al. Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension. JCI Insight 2021, 6, e148668.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885.
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414.
- Moschonas, I.C.; Tselepis, A.D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis 2019, 288, 9–16.
- Sun, S.; Zou, X.; Wang, D.; Liu, Y.; Zhang, Z.; Guo, J.; Lu, R.; Huang, W.; Wang, S.; Li, Z.; et al. IRGM/Irgm1 deficiency inhibits neutrophil-platelet interactions and thrombosis in experimental atherosclerosis and arterial injury. Biomed. Pharmacother. 2023, 158, 114152.
- Dou, H.; Kotini, A.; Liu, W.; Fidler, T.; Endo-Umeda, K.; Sun, X.; Olszewska, M.; Xiao, T.; Abramowicz, S.; Yalcinkaya, M.; et al. Oxidized Phospholipids Promote NETosis and Arterial Thrombosis in LNK(SH2B3) Deficiency. Circulation 2021, 144, 1940–1954.
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320.
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215.
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677.
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4063562.
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109.
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153.
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287.
- Feng, G.; Yang, X.; Li, Y.; Wang, X.; Tan, S.; Chen, F. LPS enhances platelets aggregation via TLR4, which is related to mitochondria damage caused by intracellular ROS, but not extracellular ROS. Cell. Immunol. 2018, 328, 86–92.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414.
- Lim, G.B. IL-1 signalling in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 200.
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289.
- Badimon, L. Interleukin-18: A potent pro-inflammatory cytokine in atherosclerosis. Cardiovasc. Res. 2012, 96, 172–175; discussion 176–180.
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603.
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The inflammasome: First line of the immune response to cell stress. Cell 2006, 126, 659–662.
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031.
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197.
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853.
- Liao, T.-L.; Chen, Y.-M.; Tang, K.-T.; Chen, P.-K.; Liu, H.-J.; Chen, D.-Y. MicroRNA-223 inhibits neutrophil extracellular traps formation through regulating calcium influx and small extracellular vesicles transmission. Sci. Rep. 2021, 11, 15676.
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689.
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9.
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Yao, M.; Liu, Y.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin. Sci. 2019, 133, 565–582.

