2. NETs, NETosis and Atherosclerosis
Serving as immune sentinels, neutrophils are the first to react against infectious agents and the first to reach the site of injury to catch and kill pathogens by phagocytosis to resolve inflammation (clearing pro-inflammatory stimuli) and repair tissue (promote angiogenesis) [
8]. A few years ago, a new anti-pathogen strategy of neutrophils was discovered whereby on meeting a pathogen, neutrophils make a mesh-like structure called a neutrophil extracellular trap (NET) that ensnares and neutralizes pathogens [
11]. NETs are web-like structures formed via decondensation of their chromatin by citrullination of arginine by peptidyl arginine deiminase 4 (PAD4) [
13]. This loose chromatin becomes embedded with azurophilic granules and cytosolic proteins. The components of decondensed chromatin include predominantly positively charged proteins along with cell-free DNA and RNA. Although 70% of the proteins are histones, the rest belongs to the cytoplasm, metabolic pathways and cytoskeleton. Almost 20 proteins have been identified in the NET proteome (NETome) that participate in NET formation. These include neutrophil elastase (NE), proteinase-3 (PR3), myeloperoxidase (MPO), Cathepsin G, Keratinocyte transglutaminase, factor XIIIa, alpha-defensins and citrullinated histones (ctH) [
16].
When neutrophils fail to resolve inflammation by phagocytosis and pro-inflammatory stimuli are non-subsiding and incessant then NETs are formed and thrown on the microbes (pro-inflammatory stimulus) either by breaking the plasma membrane with the pore-forming protein Gasdermin D (GSDMD) causing the death of the neutrophil (suicidal NETosis) or by transporting these NETs by membrane blebbing or vesicular exocytosis (vital NETosis) [
10,
13]. NETosis is the process by which the neutrophil expels its nuclear material outside the cell; however this term was earlier used for neutrophil death (
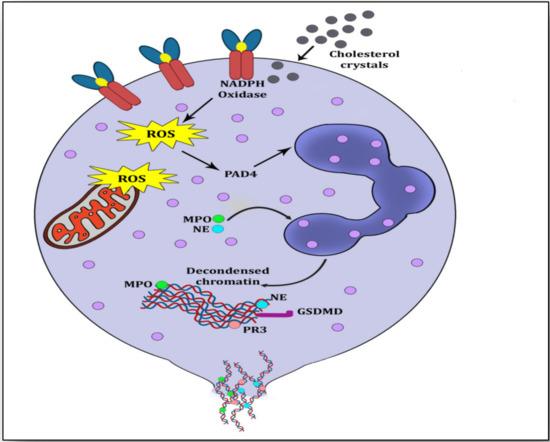
Figure 2).
Figure 2. Overview of suicidal NETosis. Cholesterol crystals interact with free radicals and generate NADPH-oxidase-induced reactive oxygen species (ROS). ROS stimulate peptidyl arginine deiminase 4 (PADI4) to citrullinate arginine resulting in loosening of chromatin from histone. Myeloperoxidase (MPO) and neutrophil elastase (NE) migrate to the nuclear membrane for its rupturing by further decondensation of the chromatin. This decondensed chromatin exhibits a mesh-like structure called neutrophil extracellular trap (NET), which is ejected into the cytoplasm, where it is embedded with azurophilic granules and cytosolic proteins. Finally, this NET is ejected through the membrane rupturing of the neutrophil and causing its death.
NET formation or NETosis is initiated by several triggers; otherwise, resting neutrophils are non-inflammatory and do not undergo NETosis [
8]. Vital NETosis is observed mostly during infection rather than sterile injury, whereas suicidal NETosis is associated with sterile and noninfectious complications [
13]. Several stimuli have been observed to initiate the formation of NETs such as phorbol-12-myristate-13-acetate (PMA) [
17], bacterial toxin; ionomycin, lipopolysaccharides (LPS) [
10], some cytokines such as IL-1β, TNFα and IL-8 [
11], microbe size [
18], activated platelets [
19], reactive oxygen species (ROS) burst [
20], histone acetylation [
21], etc.
Atherosclerosis is considered to be the chief culprit in the pathology of several complex disorders, and is propelled by vascular inflammation [
1]. The inflammatory trigger by lipid-rich foam cells in atherosclerosis is considered to be the central event when these accumulate in the subendothelial area of an injured artery [
1,
3,
5]. In order to clarify whether NETs are formed during and contribute to atherosclerosis, earlier studies have shown that neutrophils were either present with condensed nuclei or were luminar rather than lesional, suggesting that neutrophils are less likely to participate in atherosclerosis development [
22,
23]. In essential hypertension patients, abundant NET formation was observed but when they were treated with angiotensin II (AngII), NETs were substantially reduced [
24]. It has been observed that a mouse knockout for
ApoE-/- expresses heightened NET formation and interferon-alpha (IFN-α) expression in atherosclerotic arteries. When these mice were injected daily with Cl-amidine, which is an inhibitor of the PAD4 enzyme, recruitment of neutrophils and macrophages into intima was significantly reduced, hence mitigating NET formation and reducing atherosclerotic load by delaying carotid thrombosis [
22]. This suggests that PAD4 is a paramount enzyme for histone citrullination and recruitment of NETs during atherosclerosis.
NETs and NETosis not only participate in atherosclerosis but also contribute to thrombus formation. NETs induce a scaffold of DNA that exhibits a red blood cell (RBC)-rich thrombus along with von Willebrand factor (vWF), fibronectin and fibrinogen in experimental deep venous thrombus in baboons [
12]. This inference is corroborated by a study in humans showing that activated platelets interact with neutrophils to generate tissue factors that provoke neutrophils to induce thrombogenic signals promoting atherogenesis in ST-segment elevation acute myocardial infarction (STEMI) [
28]. NETs participate significantly in prothrombotic signaling by triggering the oxidation of LDLs, generating ROS, endothelial dysfunction, apoptosis, fibrin-formation-induced platelet aggregation, accumulation of vWF and fibrinogen [
29]. NETs are observed to interact with inflammatory platelets to promote thrombosis via immune-related GTPase family M protein (IRGM) and its orthologs [
30]. Carriers of the homozygote TT genotype of the R262W polymorphism within the Src homology 2B (SH2B) protein 3 (LNK/SH2B3) gene show augmented platelet–neutrophil aggregation leading to heightened atherosclerosis and atherothrombosis in an oxidized phospholipid (oxPL)-dependent manner [
31]. Alluding to contradictions and confusions, a remarkable piece of research has answered three important queries related to the role and relevance of NETs and NETosis in an experimental murine model of atherosclerosis [
32].
3. NLRP3 Inflammasome Activation and Atherosclerosis
NLRP3 is present in the cytoplasm as an inactive protein but is activated on sensing danger from several cellular triggers [
9,
35]. NLRP3 inflammasome activation has been observed to play a central role in initiating the inflammatory cascade in several diseases [
36]. It is a multiprotein complex that contains an adapter (ASC or PYCARD), a receptor (NLRP3) and an effector (pro-caspase-1) along with domains such as telomerase-associated protein 1 (TP1 or NACHT), neuronal apoptosis inhibitory protein (NAIP), N-terminal pyrin domain (PYD) and leucine-rich repeat (LRR). On sensing damage or danger signals by NACHT, it triggers signaling where ASC forms speck-like clusters and pro-caspase 1 (Pro-CASP1) is recruited to the ASC speck clusters. ASC and Pro-CASP1 cleave proteolytically the active caspase-1 (CASP-1), which matures pro-IL1β to IL-1β and pro-IL18 to IL-18. During this maturation, CASP-1 induces pyroptosis, which is a form of lytic cell death triggered by the formation of plasma membrane pores by gasdermin D, leading to a flux of ions (K
+ and Ca
2+) and releasing mature IL-1β and IL-18 into the extracellular space [
37] (
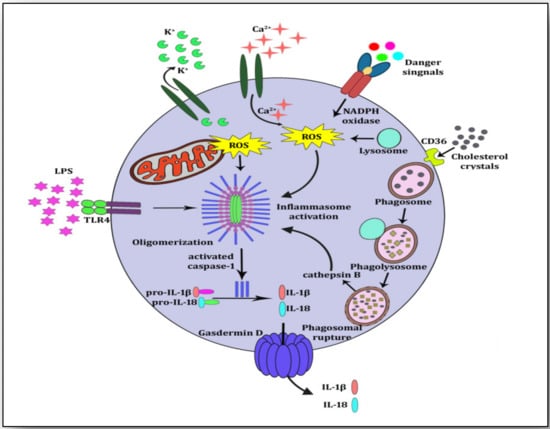
Figure 3).
Figure 3. NLRP3 inflammasome activation. Cholesterol crystals are internalized by CD36 and taken by phagosomes for phagocytosis. Cholesterol crystals are broken down and lysosomes attach to phagosomes to form phagolysosomes. Because of the size and chemistry of the cholesterol crystals, the phagolysosome ruptures and undegraded crystals along with cathepsin B are exposed in cytoplasm. This is the priming signal for NLRP3 inflammasome activation. Other signals such as K+ efflux, Ca2+ influx and lipopolysaccharide (LPS) may also trigger the NLRP3 inflammasome. Immature forms of IL-1β and IL-18 (pro-IL-1β and pro-Il-18) are proteolytically cleaved by activated caspase-1. These mature cytokines IL-1β and IL-18 are released into the extra cellular space by pore-forming Gasdermin D.
It is well known now that for the activation of the NLRP3 inflammasome, two molecular signals are required. First, a nuclear factor kappa B (NF-κB)-dependent priming signal promotes the upregulation of IL-1β and NLRP3, and then a second signal triggers the oligomerization or activation of the NLRP3 inflammasome [
38]. During atherosclerosis, cholesterol crystals evoke NETs to provide the first priming signal for macrophages, whereas the production of pro-IL-1β provides the second signal for the activation of the NLRP3 inflammasome and the release of mature cytokines. The NLRP3 inflammasome is triggered by different danger signals such as cholesterol crystals in atherosclerosis [
39], uric acid crystals in gout [
40] and amyloid-beta in Alzheimer’s [
41]. It is also activated in response to other triggers such as a reduced K
+ concentration in the cytoplasm resulting in P2X purinoceptor 4 (P2X4) receptor-mediated K
+ efflux [
42]. Necrotic cells of the ischemic core discharge Ca
2+ which increases in extracellular spaces prompting an increased influx and decreased efflux of Ca
2+ [
43]. Extensive Ca
2+ influx induces cytochrome C dislocation which impairs the mitochondrial function of ATP production leading to ROS generation [
44], which triggers NLRP3 inflammasome activation [
45].
The role of NLRP3 inflammasome activation in atherosclerosis and atherogenesis can be understood by the role of two terrible cytokines produced by it, i.e., IL-1β and IL-18. Inflammation itself does not initiate atherogenesis; rather it invites several intermediaries, especially cytokines, which mediate both inflammation and immune signaling. IL-1β plays both of these roles as a key messenger for propelling inflammatory signaling to atherothrombosis [
54].
Therefore, strategies for blocking IL-1β through inhibitors and agonists have proved to be very beneficial for atherosclerosis-driven diseases [
58]. Similarly, IL-18, another proinflammatory cytokine produced by the activation of the NLRP3 inflammasome, is expressed in macrophages and plays a significant role in inflammatory and immune signaling by the synthesis of T cells, natural killer cells and IFNγ [
59]. A study has revealed that it is rampantly present in atherosclerotic plaques and influences plaque destabilization [
60].
4. NETosis-NLRP3 Inflammasome Activation Link: A Maleficent Crosstalk
Clinical research on the signaling pathways leading to atherosclerosis has suggested that components of NETosis evoke NLRP3 inflammasome activation, which releases the proinflammatory cytokines IL-1β and IL-18. Both of these cytokines are very harmful for stimulating the atherosclerotic plaque to atherothrombosis and its rupture leading to ischemia. A study has revealed that monocyte-derived macrophages generate strong signals for inflammasome activation when they are incubated with NETs and then with cholesterol crystals [
32]. This shows that NETs prime macrophages to produce pro-IL-1β and cholesterol crystals induce phagolysosomal damage due to internalization by binding to CD36 (a glycoprotein on the plasma membrane of macrophages). When macrophages ingest cholesterol crystals as cellular debris, they destroy them into phagosomes; these phagosomes deliver the degraded contents to lysosomes (phagolysosomes) where they are further degraded by acid hydrolases. These hydrolyzed contents of cholesterol crystals may cause the rupture of the phagolysosome membrane and the release of its contents into the cytoplasm which otherwise are exported outside the cell by membrane transporters [
39,
65]. These undegraded cholesterol crystals released into the cytoplasm due to phagolysosomal membrane rupture, are taken as danger signals which are sensed by PRRs, termed danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). NLRP3 is a significant member of the innate immune system which is a PRR and is activated in response to these danger signals. During NLRP3 inflammasome activation, it transposes ASC and regulates caspase-1 (CASP1) stimulation, which further proteolytically cleaves the mature forms of the proinflammatory cytokines IL-1β from pro-IL-1β and IL-18 from pro-IL-18. Present abundantly in atheromatous lesions, both IL-1β and IL-18 help in plaque development and its progression to ischemic stroke [
66]. The consequent release of undegraded cholesterol crystals into the cytoplasm sets the wheel of inflammatory cascade (activating NLRP3 inflammasome) in motion through cycles of internalization of cholesterol crystals and continued phagolysosome rupturing (
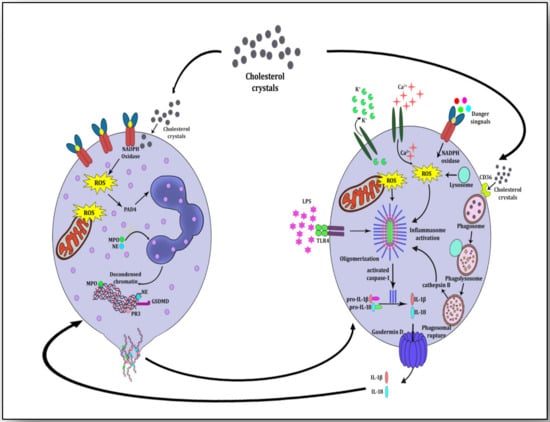
Figure 4). During acute inflammation, resolvins and selectins induce neutrophil apoptosis; they are engulfed and phagocytosed by macrophages in an effort to resolve inflammation and improve healing [
67]. Hence, it is worthwhile to infer from the experimental evidence that cholesterol crystals incite both neutrophils and macrophages to initiate pronounced inflammation (NETosis and NLRP3 inflammasome activation) and destroy the neutrophil–macrophage amity which is otherwise anti-inflammatory, pro-healing and pro-repair [
67].
Figure 4. Neutrophil-macrophage crosstalk for mutually inciting atherosclerosis and igniting inflammation. Components of NETs (dead mitochondria and cfDNA) prime macrophages and components of NLRP3 inflammasome activation (IL-1β and IL-18) induce neutrophils to form NETs. Cholesterol crystals evoke signals for both neutrophils and macrophages for the development of NETs and prime NLRP3 inflammasome by interacting with reactive oxygen species (ROS) and CD36-mediated internalization, respectively.
All of this suggests that PAD4 is required for NLRP3 inflammasome activation which propels NETosis both in vivo and in vitro under non-sterile conditions. Interestingly, IL-1β, a final product of NLRP3 inflammasome activation, is observed to play a significant role in NET formation and induces NETosis in systemic inflammatory response syndrome (SIRS) and abdominal aortic aneurysms (AAA) [
11,
70]. Moreover, cytokine IL-18 enhances the influx of Ca
2+ into neutrophils which generates mitochondrial ROS and induces NET formation [
71]. It has been demonstrated that similar processes are evident in NLRP3 inflammasome activation and in the generation of NETs [
72]. Inflammasome activation proceeds to osmotic swelling of the cells, cell necrosis and finally the release of proinflammatory IL-1β, IL-18 and some DAMPs such as interleukin 1 alpha (IL-1α), high mobility group box 1 (HMGB1) proteins and ATPs. During this process, activated CASP1/11 cleaves GSDMD which neutralizes the membrane via pore formation and eventually cause pyroptosis, an inflammation-stimulated type of cell death. A similar process is evident in neutrophils where the NLRP3 inflammasome activation triggers the cleavage of GSDMD which helps in the degradation of the granular membrane, decondensation of chromatin, dissolution of the plasma membrane and expulsion of the components of NETs [
72].
On the other hand, NET components including cfDNA stimulate NLRP3 inflammasome in macrophages [
73]. Three significant inferences are evident from the analysis of these previously mentioned research reports. First, PAD4 is the hallmark for NET generation, and it also stimulates NLRP3 inflammasome activation. Second, cholesterol crystals trigger priming and activating signals for NLRP3 inflammasome activation and also initiate NET formation and prompt NETosis. Third, components of NETs (dead mitochondria and cfDNA) prime NLRP3 to activate the inflammasome, whereas IL-1β and IL-18 stimulate neutrophils for NET formation. Although it was known that NLRP3 inflammasome activation in macrophages is responsible for delayed wound healing in diabetic mice, it has been revealed that NET production in diabetic wounds triggers NLRP3 inflammasome activation and releases IL-1β in macrophages causing a sustained inflammatory response and prolonging wound healing [
74]. Furthermore, NETs elicit signals for ROS generation triggering thioredoxin interacting protein (TXNIP) activation and both of these induce NLRP3 inflammasome activation. On summing up, one may reason to believe that both NLRP3 inflammasome activation and NETosis are triggered simultaneously, primarily by cholesterol crystals, and together these processes not only participate in triggering atherosclerosis but also perpetuate it with inflammation and exacerbate it with atherogenesis, finally leading to ischemia and infarction.