 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jacques Fantini | -- | 3487 | 2023-04-21 15:09:57 | | | |
| 2 | Jason Zhu | Meta information modification | 3487 | 2023-04-23 03:56:49 | | |
Video Upload Options
Although very different, in terms of their genomic organization, their enzymatic proteins, and their structural proteins, HIV and SARS-CoV-2 have an extraordinary evolutionary potential in common. Faced with various selection pressures that may be generated by treatments or immune responses, these RNA viruses demonstrate very high adaptive capacities, which result in the continuous emergence of variants and quasi-species. HIV and SARS-CoV-2 first recognize a lipid raft microdomain that acts as a landing strip for viral particles on the host cell surface. In the case of mucosal cells, which are the primary targets of both viruses, these microdomains are enriched in anionic glycolipids (gangliosides) forming a global electronegative field. Both viruses use lipid rafts to surf on the cell surface in search of a protein receptor able to trigger the fusion process. This implies that viral envelope proteins are both geometrically and electrically compatible to the biomolecules they select to invade host cells.
1. Introduction
2. Structural and Functional Analysis of the SARS-CoV-2 Spike Protein
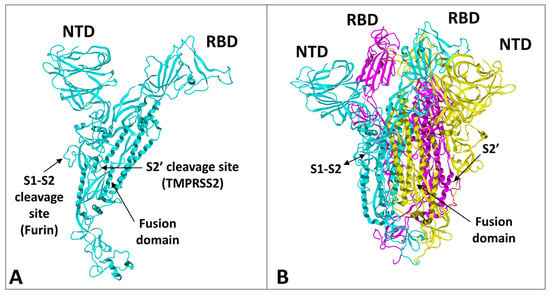
3. Structural Dynamics of SARS-CoV-2 Spike Protein Evolution
4. Structural and Functional Analysis of HIV-1 gp120 Surface Envelope Glycoprotein
5. Structural Dynamics of HIV-1 gp120 Surface Envelope Glycoprotein Evolution
6. Considerations on the Electrostatic Surface Potential
References
- Chermann, J.C.; Donker, G.; Yahi, N.; Salaun, D.; Guettari, N.; Gayet, O.; Hirsh, I. Discrepancies in AIDS virus data. Nature 1991, 351, 277–278.
- Venaud, S.; Yahi, N.; Fehrentz, J.L.; Guettari, N.; Nisato, D.; Hirsch, I.; Chermann, J.C. Inhibition of HIV by an anti-HIV protease synthetic peptide blocks an early step of viral replication. Res. Virol. 1992, 143, 311–319.
- Fantini, J.; Yahi, N.; Chermann, J.C. Human immunodeficiency virus can infect the apical and basolateral surfaces of human colonic epithelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 9297–9301.
- Yahi, N.; Baghdiguian, S.; Moreau, H.; Fantini, J. Galactosyl ceramide (or a closely related molecule) is the receptor for human immunodeficiency virus type 1 on human colon epithelial HT29 cells. J. Virol. 1992, 66, 4848–4854.
- Harouse, J.M.; Bhat, S.; Spitalnik, S.L.; Laughlin, M.; Stefano, K.; Silberberg, D.H.; Gonzalez-Scarano, F. Inhibition of entry of HIV-1 in neural cell lines by antibodies against galactosyl ceramide. Science 1991, 253, 320–323.
- Fantini, J.; Yahi, N.; Baghdiguian, S.; Chermann, J.C. Human colon epithelial cells productively infected with human immunodeficiency virus show impaired differentiation and altered secretion. J. Virol. 1992, 66, 580–585.
- Dayanithi, G.; Yahi, N.; Baghdiguian, S.; Fantini, J. Intracellular calcium release induced by human immunodeficiency virus type 1 (HIV-1) surface envelope glycoprotein in human intestinal epithelial cells: A putative mechanism for HIV-1 enteropathy. Cell Calcium 1995, 18, 9–18.
- Delézay, O.; Yahi, N.; Tamalet, C.; Baghdiguian, S.; Boudier, J.A.; Fantini, J. Direct effect of type 1 human immunodeficiency virus (HIV-1) on intestinal epithelial cell differentiation: Relationship to HIV-1 enteropathy. Virology 1997, 238, 231–242.
- Fantini, J.; Maresca, M.; Hammache, D.; Yahi, N.; Delézay, O. Glycosphingolipid (GSL) microdomains as attachment platforms for host pathogens and their toxins on intestinal epithelial cells: Activation of signal transduction pathways and perturbations of intestinal absorption and secretion. Glycoconj. J. 2000, 17, 173–179.
- Maresca, M.; Mahfoud, R.; Garmy, N.; Kotler, D.P.; Fantini, J.; Clayton, F. The virotoxin model of HIV-1 enteropathy: Involvement of GPR15/Bob and galactosylceramide in the cytopathic effects induced by HIV-1 gp120 in the HT-29-D4 intestinal cell line. J. Biomed. Sci. 2003, 10, 156–166.
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20.
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775.
- Negi, G.; Sharma, A.; Dey, M.; Dhanawat, G.; Parveen, N. Membrane attachment and fusion of HIV-1, influenza A, and SARS-CoV-2: Resolving the mechanisms with biophysical methods. Biophys. Rev. 2022, 14, 1109–1140.
- Sasaki, M.; Uemura, K.; Sato, A.; Toba, S.; Sanaki, T.; Maenaka, K.; Hall, W.W.; Orba, Y.; Sawa, H. SARS-CoV-2 variants with mutations at the S1/S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLoS Pathog. 2021, 17, e1009233.
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-Mediated Entry via the Endosome of Human Coronavirus 229E. J. Virol. 2009, 83, 712–721.
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263.
- Fantini, J.; Chahinian, H.; Yahi, N. Leveraging coronavirus binding to gangliosides for innovative vaccine and therapeutic strategies against COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 132–136.
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function and role in HIV, Alzheimer’s and prion diseases. Expert Rev. Mol. Med. 2002, 4, 1–22.
- Fantini, J.; Yahi, N.; Azzaz, F.; Chahinian, H. Structural dynamics of SARS-CoV-2 variants: A health monitoring strategy for anticipating Covid-19 outbreaks. J. Infect. 2021, 83, 197–206.
- Sun, X.L. The role of cell surface sialic acids for SARS-CoV-2 infection. Glycobiology 2021, 31, 1245–1253.
- Palacios-Rápalo, S.N.; De Jesús-González, L.A.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Osuna-Ramos, J.F.; Martínez-Mier, G.; Quistián-Galván, J.; Muñoz-Pérez, A.; Bernal-Dolores, V.; Del Ángel, R.M.; et al. Cholesterol-Rich Lipid Rafts as Platforms for SARS-CoV-2 Entry. Front. Immunol. 2021, 12, 796855.
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.A.; Ghosh, S.; et al. The structural basis of accelerated host cell entry by SARS-CoV-2. FEBS J. 2021, 288, 5010–5020.
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 55, 105960.
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278.
- Pak, A.J.; Yu, A.; Ke, Z.; Briggs, J.A.G.; Voth, G.A. Cooperative multivalent receptor binding promotes exposure of the SARS-CoV-2 fusion machinery core. Nat. Commun. 2022, 13, 1002.
- Guérin, P.; Yahi, N.; Azzaz, F.; Chahinian, H.; Sabatier, J.M.; Fantini, J. Structural Dynamics of the SARS-CoV-2 Spike Protein: A 2-Year Retrospective Analysis of SARS-CoV-2 Variants (from Alpha to Omicron) Reveals an Early Divergence between Conserved and Variable Epitopes. Molecules 2022, 27, 3851.
- Colson, P.; Lavagna, C.; Delerce, J.; Groshenry, G.; Yahi, N.; Fantini, J.; La Scola, B.; Althaus, T. First Detection of the SARS-CoV-2 Omicron BA.5/22B in Monaco. Microorganisms 2022, 10, 1952.
- Yahi, N.; Chahinian, H.; Fantini, J. Infection-enhancing anti-SARS-CoV-2 antibodies recognize both the original Wuhan/D614G strain and Delta variants. A potential risk for mass vaccination? J. Infect. 2021, 83, 607–635.
- Al-Khatib, H.A.; Smatti, M.K.; Ali, F.H.; Zedan, H.T.; Thomas, S.; Ahmed, M.N.; El-Kahlout, R.A.; Al Bader, M.A.; Elgakhlab, D.; Coyle, P.V.; et al. Comparative analysis of within-host diversity among vaccinated COVID-19 patients infected with different SARS-CoV-2 variants. iScience 2022, 25, 105438.
- Azzaz, F.; Yahi, N.; Di Scala, C.; Chahinian, H.; Fantini, J. Ganglioside binding domains in proteins: Physiological and pathological mechanisms. Adv. Protein Chem. Struct. Biol. 2022, 128, 289–324.
- Gowrisankar, A.; Priyanka, T.M.C.; Banerjee, S. Omicron: A mysterious variant of concern. Eur. Phys. J. Plus 2022, 137, 100.
- Mahase, E. Omicron: South Africa says fourth wave peak has passed as it lifts curfew. BMJ Br. Med. J. 2022, 376, o7.
- Haschka, T.; Vergu, E.; Roche, B.; Poletto, C.; Opatowski, L. Retrospective analysis of SARS-CoV-2 omicron invasion over delta in French regions in 2021-22: A status-based multi-variant model. BMC Infect. Dis. 2022, 22, 815.
- Bal, A.; Simon, B.; Destras, G.; Chalvignac, R.; Semanas, Q.; Oblette, A.; Quéromès, G.; Fanget, R.; Regue, H.; Morfin, F.; et al. Detection and prevalence of SARS-CoV-2 co-infections during the Omicron variant circulation in France. Nat. Commun. 2022, 13, 6316.
- Grabowski, F.; Kochańczyk, M.; Lipniacki, T. The Spread of SARS-CoV-2 Variant Omicron with a Doubling Time of 2.0-3.3 Days Can Be Explained by Immune Evasion. Viruses 2022, 14, 294.
- Zhang, L.; Li, Q.; Liang, Z.; Li, T.; Liu, S.; Cui, Q.; Nie, J.; Wu, Q.; Qu, X.; Huang, W. The significant immune escape of pseudotyped SARS-CoV-2 variant Omicron. Emerg. Microbes Infect. 2022, 11, 1–5.
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179.
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.-L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.-M.; Chu, A.W.-H.; Chan, K.-H.; et al. SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg. Microbes Infect. 2022, 11, 277–283.
- Willett, B.J.; Grove, J.; MacLean, O.; Wilkie, C.; Logan, N.; De Lorenzo, G.; Furnon, W.; Scott, S.; Manali, M.; Szemiel, A. The hyper-transmissible SARS-CoV-2 Omicron variant exhibits significant antigenic change, vaccine escape and a switch in cell entry mechanism. MedRxiv 2022.
- Fantini, J.; Yahi, N.; Colson, P.; Chahinian, H.; La Scola, B.; Raoult, D. The puzzling mutational landscape of the SARS-2-variant Omicron. J. Med. Virol. 2022, 94, 2019–2025.
- Jaafar, R.; Boschi, C.; Aherfi, S.; Bancod, A.; Le Bideau, M.; Edouard, S.; Colson, P.; Chahinian, H.; Raoult, D.; Yahi, N.; et al. High Individual Heterogeneity of Neutralizing Activities against the Original Strain and Nine Different Variants of SARS-CoV-2. Viruses 2021, 13, 2177.
- Akaishi, T.; Fujiwara, K.; Ishii, T. Insertion/deletion hotspots in the Nsp2, Nsp3, S1, and ORF8 genes of SARS-related coronaviruses. BMC Ecol. Evol. 2022, 22, 123.
- Tamalet, C.; Izopet, J.; Koch, N.; Fantini, J.; Yahi, N. Stable rearrangements of the beta3-beta4 hairpin loop of HIV-1 reverse transcriptase in plasma viruses from patients receiving combination therapy. AIDS 1998, 12, F161–F166.
- Tamalet, C.; Yahi, N.; Tourrès, C.; Colson, P.; Quinson, A.M.; Poizot-Martin, I.; Dhiver, C.; Fantini, J. Multidrug resistance genotypes (insertions in the beta3-beta4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: Incidence and association with other resistance mutations. Virology 2000, 270, 310–316.
- Li, Z.; Li, W.; Lu, M.; Bess, J., Jr.; Chao, C.W.; Gorman, J.; Terry, D.S.; Zhang, B.; Zhou, T.; Blanchard, S.C.; et al. Subnanometer structures of HIV-1 envelope trimers on aldrithiol-2-inactivated virus particles. Nat. Struct. Mol. Biol. 2020, 27, 726–734.
- Willey, R.L.; Bonifacino, J.S.; Potts, B.J.; Martin, M.A.; Klausner, R.D. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. USA 1988, 85, 9580–9584.
- Chatterjee, S.; Basak, S.; Khan, N.C. Morphogenesis of human immunodeficiency virus type 1. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 1992, 60, 181–186.
- Turner, B.G.; Summers, M.F. Structural biology of HIV. J. Mol. Biol. 1999, 285, 1–32.
- Fantini, J.; Chahinian, H.; Yahi, N. A Vaccine Strategy Based on the Identification of an Annular Ganglioside Binding Motif in Monkeypox Virus Protein E8L. Viruses 2022, 14, 2531.
- Hammache, D.; Yahi, N.; Piéroni, G.; Ariasi, F.; Tamalet, C.; Fantini, J. Sequential interaction of CD4 and HIV-1 gp120 with a reconstituted membrane patch of ganglioside GM3: Implications for the role of glycolipids as potential HIV-1 fusion cofactors. Biochem. Biophys. Res. Commun. 1998, 246, 117–122.
- Hug, P.; Lin, H.M.; Korte, T.; Xiao, X.; Dimitrov, D.S.; Wang, J.M.; Puri, A.; Blumenthal, R. Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J. Virol. 2000, 74, 6377–6385.
- Popik, W.; Alce, T.M.; Au, W.C. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J. Virol. 2002, 76, 4709–4722.
- Sorice, M.; Garofalo, T.; Misasi, R.; Longo, A.; Mattei, V.; Sale, P.; Dolo, V.; Gradini, R.; Pavan, A. Evidence for cell surface association between CXCR4 and ganglioside GM3 after gp120 binding in SupT1 lymphoblastoid cells. FEBS Lett. 2001, 506, 55–60.
- Nguyen, D.H.; Giri, B.; Collins, G.; Taub, D.D. Dynamic reorganization of chemokine receptors, cholesterol, lipid rafts, and adhesion molecules to sites of CD4 engagement. Exp. Cell Res. 2005, 304, 559–569.
- Carter, G.C.; Bernstone, L.; Sangani, D.; Bee, J.W.; Harder, T.; James, W. HIV entry in macrophages is dependent on intact lipid rafts. Virology 2009, 386, 192–202.
- Campbell, S.M.; Crowe, S.M.; Mak, J. Lipid rafts and HIV-1: From viral entry to assembly of progeny virions. J. Clin. Virol. 2001, 22, 217–227.
- Mañes, S.; del Real, G.; Lacalle, R.A.; Lucas, P.; Gómez-Moutón, C.; Sánchez-Palomino, S.; Delgado, R.; Alcamí, J.; Mira, E.; Martínez, A.C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000, 1, 190–196.
- Kozak, S.L.; Heard, J.M.; Kabat, D. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J. Virol. 2002, 76, 1802–1815.
- Fragoso, R.; Ren, D.; Zhang, X.; Su, M.W.; Burakoff, S.J.; Jin, Y.J. Lipid raft distribution of CD4 depends on its palmitoylation and association with Lck, and evidence for CD4-induced lipid raft aggregation as an additional mechanism to enhance CD3 signaling. J. Immunol. 2003, 170, 913–921.
- Sorice, M.; Garofalo, T.; Misasi, R.; Longo, A.; Mikulak, J.; Dolo, V.; Pontieri, G.M.; Pavan, A. Association between GM3 and CD4-Ick complex in human peripheral blood lymphocytes. Glycoconj. J. 2000, 17, 247–252.
- Sattentau, Q.J.; Moore, J.P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415.
- Pinter, A.; Honnen, W.J.; Tilley, S.A. Conformational changes affecting the V3 and CD4-binding domains of human immunodeficiency virus type 1 gp120 associated with env processing and with binding of ligands to these sites. J. Virol. 1993, 67, 5692–5697.
- Hammache, D.; Yahi, N.; Maresca, M.; Piéroni, G.; Fantini, J. Human erythrocyte glycosphingolipids as alternative cofactors for human immunodeficiency virus type 1 (HIV-1) entry: Evidence for CD4-induced interactions between HIV-1 gp120 and reconstituted membrane microdomains of glycosphingolipids (Gb3 and GM3). J. Virol. 1999, 73, 5244–5248.
- Hammache, D.; Piéroni, G.; Yahi, N.; Delézay, O.; Koch, N.; Lafont, H.; Tamalet, C.; Fantini, J. Specific interaction of HIV-1 and HIV-2 surface envelope glycoproteins with monolayers of galactosylceramide and ganglioside GM3. J. Biol. Chem. 1998, 273, 7967–7971.
- Rawat, S.S.; Johnson, B.T.; Puri, A. Sphingolipids: Modulators of HIV-1 infection and pathogenesis. Biosci. Rep. 2005, 25, 329–343.
- Cormier, E.G.; Dragic, T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 2002, 76, 8953–8957.
- Gallo, S.A.; Finnegan, C.M.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2003, 1614, 36–50.
- Richman, D.D.; Bozzette, S.A. The Impact of the Syncytium-Inducing Phenotype of Human Immunodeficiency Virus on Disease Progression. J. Infect. Dis. 1994, 169, 968–974.
- Rosen, O.; Sharon, M.; Quadt-Akabayov, S.R.; Anglister, J. Molecular switch for alternative conformations of the HIV-1 V3 region: Implications for phenotype conversion. Proc. Natl. Acad. Sci. USA 2006, 103, 13950–13955.
- Kuiken, C.L.; De Jong, J.; Baan, E.; Keulen, W.; Tersmette, M.; Goudsmit, J. Evolution of the V3 envelope domain in proviral sequences and isolates of human immunodeficiency virus type 1 during transition of the viral biological phenotype. J. Virol. 1992, 66, 4622–4627.
- Xiao, L.; Owen, S.M.; Goldman, I.; Lal, A.A.; deJong, J.J.; Goudsmit, J.; Lal, R.B. CCR5 Coreceptor Usage of Non-Syncytium-Inducing Primary HIV-1 Is Independent of Phylogenetically Distinct Global HIV-1 Isolates: Delineation of Consensus Motif in the V3 Domain That Predicts CCR-5 Usage. Virology 1998, 240, 83–92.
- Björndal, A.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyö, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487.
- Balasubramanian, C.; Chillemi, G.; Abbate, I.; Capobianchi, M.R.; Rozera, G.; Desideri, A. Importance of V3 loop flexibility and net charge in the context of co-receptor recognition. A molecular dynamics study on HIV gp120. J. Biomol. Struct. Dyn. 2012, 29, 879–891.
- Repits, J.; Sterjovski, J.; Badia-Martinez, D.; Mild, M.; Gray, L.; Churchill, M.J.; Purcell, D.F.; Karlsson, A.; Albert, J.; Fenyö, E.M. Primary HIV-1 R5 isolates from end-stage disease display enhanced viral fitness in parallel with increased gp120 net charge. Virology 2008, 379, 125–134.
- Kalinina, O.V.; Pfeifer, N.; Lengauer, T. Modelling binding between CCR5 and CXCR4 receptors and their ligands suggests the surface electrostatic potential of the co-receptor to be a key player in the HIV-1 tropism. Retrovirology 2013, 10, 130.
- López de Victoria, A.; Kieslich, C.A.; Rizos, A.K.; Krambovitis, E.; Morikis, D. Clustering of HIV-1 subtypes based on gp120 V3 loop electrostatic properties. BMC Biophys. 2012, 5, 3.
- Shioda, T.; Levy, J.A.; Cheng-Mayer, C. Small amino acid changes in the V3 hypervariable region of gp120 can affect the T-cell-line and macrophage tropism of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1992, 89, 9434–9438.
- Clayton, F.; Kotler, D.P.; Kuwada, S.K.; Morgan, T.; Stepan, C.; Kuang, J.; Le, J.; Fantini, J. Gp120-induced Bob/GPR15 activation: A possible cause of human immunodeficiency virus enteropathy. Am. J. Pathol. 2001, 159, 1933–1939.
- Brenneman, D.E.; Westbrook, G.L.; Fitzgerald, S.P.; Ennist, D.L.; Elkins, K.L.; Ruff, M.R.; Pert, C.B. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature 1988, 335, 639–642.
- Yokoyama, M.; Naganawa, S.; Yoshimura, K.; Matsushita, S.; Sato, H. Structural dynamics of HIV-1 envelope Gp120 outer domain with V3 loop. PloS ONE 2012, 7, e37530.


