1. Introduction
Virus-cell interactions during the early stages of the infection cycle of enveloped viruses have been the subject of numerous studies for several decades. The COVID-19 pandemic has mobilized the efforts of the scientific community around the world. Among these researchers, two (N.Y. and J.F.) extensively studied the molecular mechanisms associated with the infection of human cells by HIV-1 and other retroviruses. Nouara Yahi began her work on HIV by joining, in 1988, the retrovirus laboratory newly created in Marseille by Jean-Claude Chermann, one of the co-discoverers of the AIDS virus at the Pasteur Institute in Paris. She was in charge of HIV strain isolation [
1] and antiviral drug testing [
2]. Jacques Fantini joined the team in 1990. The two then started to study the infection of human epithelial intestinal cells by HIV-1 [
3], a project which would eventually solve two issues: (i) the identification of galactosylceramide (GalCer) as an alternative HIV-1 receptor in these CD4-negative cells [
4], shortly after the group of F. Gonzalez-Scarano in Philadelphia identified it as the portal of entry of HIV-1 in neural cells [
5]; (ii) a virotoxin-induced signal transduction pathway accounting for the puzzling HIV-associated enteropathy: HIV-1 surface envelope glycoprotein gp120 binding GalCer, intracellular Ca
2+ release, microtubule disruption, and impaired absorption functions [
6,
7,
8,
9,
10].
2. Structural and Functional Analysis of the SARS-CoV-2 Spike Protein
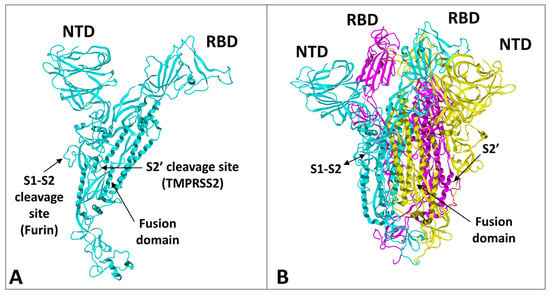
The spike protein of SARS-CoV-2 is a large glycoprotein synthesized as a precursor containing 1273 amino acid residues for the original Wuhan strain (
https://www.uniprot.org/uniprotkb/P0DTC2/entry#sequences accessed on 15 November 2022). The first 18 amino acids form the signal sequence, which is cleaved to generate the mature form encompassing residues 19-1273. As shown in
Figure 1A, the protein has a typical Y shape, whose upper branches correspond to the N-terminal domain (NTD) and the receptor-binding domain (RBD). The lower trunk of the Y displays two proteolytic sites: (i) S1–S2, which is cleaved by furin in the Golgi apparatus during biosynthesis and maturation in the infected cell, and (ii) S2′, an additional cleavage site that is essential for the virus to fuse at the plasma membrane of human lung cells [
48,
49]. The cleavage of S1–S2 by furin generates two subunits (S1 and S2) that remain non covalently attached. The S2′ site is cleaved by type II transmembrane serine protease 2 (TMPRSS2) at the cell surface or by cathepsins in the endosome [
44,
48]. In the absence of the furin site, or if the mutations render this site non-functional [
50], the alternative for the virus is, thus, to gain entry into the cell by endocytosis, according to a classic mechanism shared by many coronaviruses [
48,
51]. The S1 subunit contains the NTD and the RBD, which bind to the cell surface, whereas the S2 subunit possesses the machinery necessary for the fusion between the virus envelope and the plasma membrane of the host cell [
52]. The RBDs and the fusion machinery cluster in the center of the trimer, while the NTDs are pushed to the sides. There are two forms of the trimeric spike, the closed form and the open form [
52]. The closed form (
Figure 1B) must undergo a conformational change to make the central RBDs accessible and, thus, allow them to interact with a cellular receptor, primarily ACE2 [
53]. Therefore, the attachment of the spike protein to the ACE2 receptor cannot be the first step in the process of adhesion of the virus to the surface of the host cell [
27]. It is obvious that it is, indeed, the closed form of the spike that must first attach itself to the cell, which will trigger the conformational change, which will subsequently allow attachment to ACE2. Researchers compared this mechanism to docking a spacecraft on a space station [
27]. First, each NTD domain seeks a favorable landing area that researchers have identified as a lipid raft, i.e., a plasma membrane microdomain enriched in gangliosides and cholesterol [
17]. Indeed, researchers have shown that NTD has an excellent affinity for GM1 gangliosides, including the acetylated GM1 derivatives, which are particularly abundant on the surface of respiratory mucosal cells [
34]. Since there are 3 NTDs per spike trimer, this multiplies the chances of adhesion to a lipid raft, which ensures the initial attachment of the virus to the plasma membrane of the target cell [
54]. The first trimer sticks to a raft, causing a local deformation of the membrane, which invaginates. This allows other virus trimers to interact with vicinal rafts. The virus will then sail on these rafts until it encounters the ACE2 receptor, which is, itself, a raft-associated protein [
55]. This fast “surfing” process [
56] facilitates the formation of a trimolecular complex consisting of ACE2, raft gangliosides, and the spike protein [
29]. The next step is a conformational change of the spike trimer, which triggers the unmasking of the RBD [
57]. Finally, the spike protein is cleaved at the S2′ site by TMPRSS2 [
58], which in turn initiates the fusion process controlled by the S2 subunit [
59]. Researchers are, thus, dealing with a perfectly controlled thermodynamic mechanism, which leads inexorably from adhesion to fusion. The selection pressure that controls the evolution of SARS-CoV-2 variants at the level of the spike protein will, therefore, apply to each of these steps by independently targeting the parts of the spike protein involved. However, the parameters on which these selection pressures will act will not be the same, depending on the targeted domain. In the case of the NTD, which controls the initial step of adhesion of the virus to a lipid raft, the selection pressure will be exerted preferentially on the kinetics of adhesion. For the RBD, there will be a combination of direct effects and indirect effects. The direct effects will concern the kinetics of the RBD-ACE2 interaction, but also the affinity parameter [
34]. The indirect effects will be caused by mutations facilitating conformational unmasking of the RBD, such as the D614G mutation [
39]. However, the other mutations may, on the contrary, stabilize the trimer in its closed conformation, thus increasing the resistance of the virus to extreme conditions and favoring routes of contamination other than via the respiratory mucosa (for example by ingestion) [
60]. The furin site may be modulated by mutations facilitating proteolytic cleavage, or instead, minimizing it, in the case of the Omicron variants. Finally, mutations in the helix domains involved in the fusion mechanism may also affect the infectivity of the virus. Yet, the analysis of SARS-CoV-2 mutations is further complicated by two parameters that must also be taken into account. First, each mutation cannot be analyzed independently of other mutations in the same functional domain [
60]. Thus, the impact of a single mutation will depend not only on its own effect on the structure of the protein, but also on its contribution to a global mutational pattern. Secondly, it must be considered that, in many countries, the anti-COVID-19 vaccination rate is very high, which means that the vaccine must also be considered as a selection pressure [
61]. The recent finding of a higher intra-host diversity among vaccinated individuals is also in favor of a potential vaccine-induced immune pressure [
35]. Thus, many mutations are selected not on the basis of the advantage they confer, in terms of virus infectivity, but rather for their contribution to immune escape. People infected several times by the virus are also affected by this phenomenon, whether they are vaccinated or not.
Figure 1. Structural features of SARS-CoV-2 spike protein monomer (A) and pre-fusion trimer (B). NTD, N-terminal domain; RBD, receptor binding domain. In the trimer, the subunits ribbons are colored in cyan, yellow, and magenta. The two proteolytic cleavage sites (S1–S2 and S2′) are indicated in the monomer and (when visible) in the trimer. The models were modified from pdb file 7 bnm.
3. Structural Dynamics of SARS-CoV-2 Spike Protein Evolution
Schematically, the NTD has two fundamental properties that explain why this domain is particularly efficient in targeting the gangliosides of the lipid rafts: (i) its contact surface with the cell is flat [
29,
56], and (ii) it is globally electropositive [
34]. This combination of geometrical and electrostatic parameters is truly the winning formula for binding to lipid rafts, which are well-demarcated flat and electronegative landing zones [
62]. Each GM1 ganglioside has an ionized sialic acid and, therefore, has a negative charge at physiological pH. The repetition of this negative charge over the raft gives these membrane domains a global negative electrostatic potential [
62]. Under these conditions, the more the NTD is electropositive, the faster it will interact with the raft. Mutations that increase the electropositivity of the NTD, therefore, directly affect the kinetics of interaction of the virus with the raft [
34]. In the order of appearance of SARS-CoV-2 variants, from the original Wuhan strain to the Delta variant, the electrostatic potential underwent a steady increase, becoming more and more electropositive [
34]. However, the electrostatic potential cannot increase indefinitely because, beyond a certain value, the virus could stick too strongly to the membrane, which would have the effect of making it non-infectious. Therefore, it was clear that the Delta variant was the final outcome and no virus could supplant it, except to go back and compensate for the decrease in surface potential by another parameter. This is how the first Omicron variant appeared, when Delta was still predominant [
63]. It remained so in several countries, especially those where vaccination was the least widespread, such as South Africa [
64]. Researchers can clearly see that the analysis of variants is multiparametric and that it is sometimes necessary to look elsewhere, other than in the virus itself, to find the reasons explaining the emergence of one variant compared to another. Thus, it is in the most vaccinated countries, such as France or Denmark, that the wave of Omicron was the strongest, even though Delta was still very present [
65,
66,
67]. It is, therefore, most likely due to an immune escape phenomenon that the first Omicron variant was able to succeed Delta, with the neutralizing antibodies induced by the vaccine being notably less effective against Omicron, compared to Delta [
68,
69]. A very strong argument in favor of this interpretation is given by the in vitro infection kinetics comparing the Delta and Omicron in Calu-3 cells, which have high level expressions of TMPRSS2 and in (TMPRSS2)-overexpressing VeroE6 cells [
70]. In a competition assay with these cells, Delta outcompeted Omicron [
70]. This means that, on a strictly virological level, Omicron has no decisive advantage over Delta. For Omicron to take over Delta, it needed outside help, the natural or vaccinal immune response. However, the selection pressures are multiple and Omicron has acquired a faculty that Delta did not have, nor the other variants that preceded it: an inoperative furin site (S1–S2) condemning the virus to enter the cells through endocytosis [
70,
71]. This resistance to furin cleavage is manifested at the level of the cleavage zone by a structuring that reduces the flexibility of the substrate loop.
Finally, Omicron’s mutational program is really a jigsaw puzzle, with mutations that seem to go all over the place [
72]. The electropositive surface potential of the NTD is diminished, compared to Delta, but that of the RBD is increased. The contact surface of the NTD is slightly rounded, but this problem will be solved by successive Omicron variants, from BA.2 to BA.5 and BQ.1.1 today.
In order to provide a comparative analysis of SARS-CoV-2 variants, researchers have proposed a transmissibility index (T-index) taking into account the kinetic parameters (surface electrostatic potential of NTD and RBD) and affinity (NTD-gangliosides and RBD-ACE2 interactions) [
34,
72,
73]. This index clearly accounts for the evolution of SARS-CoV-2 from the original Wuhan strain to the Delta variant, with each new variant having a higher T-index than its predecessor. The arrival of Omicron has changed the situation, since this new line of variants has a lower T-index than Delta, with its success being due both to its mechanism of entry by endocytosis (mutations of the furin site) and to the immune escape.
One of the most intriguing aspects of the structural dynamics of the evolution of SARS-CoV-2 variants is the relative rarity of insertions and deletions, which are, nevertheless, the hallmark of RNA viruses, including HIV [
74,
75,
76]. In fact, these events also exist for SARS-CoV-2, but they are concentrated in hot spots and, in particular, at the level of the NTD [
74] and, more precisely, at the level of the contact surface with the rafts [
34]. One can wonder about the significance of these rearrangements in these zones of interaction with lipid rafts. If researchers consider the selection pressure induced by the neutralizing antibodies recognizing these areas, the immune escape requires mutations decreasing the affinity of these antibodies for the NTD [
73]. However, ultimately, these neutralizing antibodies are kind of mirrors of the NTD’s contact surface: geometrically flat and electrostatically negatively charged, similar to rafts [
27]. Under these conditions, any modification of the epitopes recognized by these antibodies could also cause a decrease of the affinity of the NTD for the rafts and, therefore, a disadvantage for the virus. The deletions in the NTD can then be interpreted as a loss of epitope, without major consequences for the binding to the rafts. The virus becomes resistant to neutralizing antibodies, while remaining infectious.
Despite the complexity of the selection pressures that determine the emergence of SARS-CoV-2 variants, the mechanisms affected by the mutations of the spike protein remain perfectly explainable. Among these mechanisms, the surface electrostatic potential plays a major role, which is, in fact, shared by many viruses, such as HIV.
4. Structural and Functional Analysis of HIV-1 gp120 Surface Envelope Glycoprotein
In the case of HIV-1, the molecular details of the mechanisms of entry are different, but the strategy is globally similar to the one used by SARS-CoV-2. The surface envelope glycoproteins that constitute the trimeric spike of HIV-1 [
77] are already cleaved from a unique precursor, gp160 [
78]. This precursor is cleaved by a cellular protease before the assembly of the viral particle at the plasma membrane of the infected cells [
79]. Two glycoproteins are generated, the surface envelope g120 (SU), which is equivalent to S1 for SARS-CoV-2, and the transmembrane gp41 (TM), which is equivalent to S2 [
80]. The amino acid residues that are critical for the binding to raft gangliosides (V3 loop for HIV-1 and part of the NTD for SARS-CoV-2) are highlighted in yellow. Although the binding to lipid rafts is typically an induced fit mechanism [
81], a significant part of the amino acid chain is exposed on the surface of the viral glycoproteins, allowing for fast contact with raft gangliosides. The sequence of events leading to HIV-1 entry is, however, very similar to the fusion process of SARS-CoV-2. Researchers elucidated and published this mechanism as early as 1998 [
19], before it was confirmed by many subsequent studies [
14,
82,
83,
84,
85,
86,
87,
88]. The HIV-1 virion first binds to a raft, via its gp120 V3 loop domain, which, although not totally exposed on the surface of the viral glycoprotein, is sufficiently accessible to engage functional virus-raft contacts. Since the CD4 receptor is also located in a raft [
89,
90], this very first step facilitates gp120-CD4 binding. A conformational change then totally unmasks the V3 loop of gp120 [
91,
92]. The V3 loop keeps the virus firmly attached to the raft through direct interactions with accessory glycosphingolipids (Gb3 and/or GM3) [
11,
13,
15,
19]. The virus will then sail on the cell surface (surfing step) [
11,
15,
87] until it encounters a co-receptor, CCR5 or CXCR4, also recognized by the V3 loop [
93]. The V3-coreceptor interaction disconnects CD4 from the complex and triggers the last conformational change of gp41, which eventually activates the fusion machinery [
94]. As in the case of SARS-CoV-2, infection can only occur if the rafts are fully functional and free to move on the cell surface. An important specificity of HIV-1 concerns the choice of the coreceptor, CCR5 or CXCR4. Initially, HIV-1 were classified according to a phenotypic criterion and the ability to induce syncytia in cultures of infected cells [
95]. The syncytium-inducing (SI) viruses appear in infected patients after the non-syncytium-inducing (NSI) viruses from which they derive by accumulation of mutations at the level of the V3 loop [
96]. The more aggressive a virus is, the more syncytia it forms and the more mutations it presents in its V3 loop [
97]. In general, NSI viruses recognize CCR5 [
98] and SI viruses recognize CXCR4 [
99]. Interestingly, this coreceptor switch is strongly correlated with an increase in the surface electrostatic potential of the V3 domain [
100,
101]. This increase is consistent with the fact that the surface potential of CXCR4 is much more electronegative than that of CCR5 [
102].
5. Structural Dynamics of HIV-1 gp120 Surface Envelope Glycoprotein Evolution
It, therefore, appears that the evolution of SARS-CoV-2 and HIV-1 variants and quasi-species is driven by an adaptation of the contact surfaces of the virus with the areas and membrane receptors controlling the adhesion of viral particles to target cells. In fact, the analysis of the evolution of the V3 loop of HIV-1 gp120 shows an accumulation of basic amino acid residues (lysine and arginine), which increase the electropositivity of this domain [
103]. It is this evolution of the surface potential that allows the virus to sequentially use the CCCR5 then CXCR4 co-receptors, while maintaining a good attractiveness for lipid rafts. From this point of view, the virus behaves like an evolutionary probe, capable of estimating the electrostatic potential of its targets and of making its own potential evolve, according to this parameter. Interestingly, this important feature was established several years before HIV-1 coreceptors were identified [
104]. Indeed, the evolution of the V3 loop sequences shows a progressive enrichment in basic amino acids. As a result, a strong correlation can be established between the net charge of the V3 loop and the type of coreceptor used. Below a net charge of +3, CCR5 will be preferred. Above the value +4, the virus will then be able to use CXCR4 [
100]. Viruses able to use both CCR5 and CXCR4 (referred to as R5X 4 strains) have an intermediary net charge in the 3–4 range [
100].
At the phenotypic level, quasi-species using CXCR4 are more aggressive, and they can infect cellular targets, other than T4 lymphocytes and macrophages. However, since secreted gp120 has virotoxin properties [
10], infection is not requested to induce deleterious effects in the intestinal epithelium [
105] and nerve cells [
106], through direct binding of the viral glycoprotein to cell surface glycosphingolipids, such as galactosylceramide [
4,
5].
6. Considerations on the Electrostatic Surface Potential
As discussed above, the surface electrostatic potential plays a major role in the selection mechanisms that are directly responsible for the emergence of new viral populations with increased tropism and/or infectivity. At this point, researchers must make an important distinction between kinetic effects and affinity enhancement [
34]. Regarding the lipid raft recognition domains, it is clear that the increase in surface electrostatic potential does not translate into an increase in the affinity of viruses for gangliosides. There are several reasons for this. First, the interaction of viral glycoproteins with raft gangliosides involves several distinct gangliosides [
62]. It is, therefore, possible to measure an overall avidity for this cluster of gangliosides, which is the sum of the affinity of each individual ganglioside for the viral glycoprotein. The variation in free energy (ΔG) associated with this multi-partner reaction is already very large, and it can no longer increase significantly [
34]. Indeed, in the case of SARS-CoV-2, this ΔG shows very little variation from one variant to another, despite the accumulation of mutations in the spike protein. In contrast, the increase in surface potential gives a clear kinetic advantage, allowing the most electropositive viruses to adhere to target cells faster than their competitors.
This advantage is conferred by the particular associations of mutations concentrated in the V3 loop of HIV-1 gp120 [
107] and at the surface of the NTD and of the RBD in the case of the spike protein of SARS-CoV-2 [
34]. In the latter case, the NTD and the RBD can evolve in concert by increasing their surface electrostatic potential, which can be visualized when observing the upper face of the spike protein trimer. This is the case for all SARS-CoV-2 variants, from the original Wuhan strain to the Delta variant, but not for the Omicron series. This series of variants differs significantly from their predecessors because, for them, the increase in surface potential essentially concerns the RBD. This unexpected asymmetry explains why the T-index of the first Omicron variant is lower than that of the last Delta variant [
72].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24031923