 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander Shpakov | -- | 8645 | 2023-04-19 21:58:16 | | | |
| 2 | Peter Tang | -3 word(s) | 8642 | 2023-04-20 04:03:12 | | |
Video Upload Options
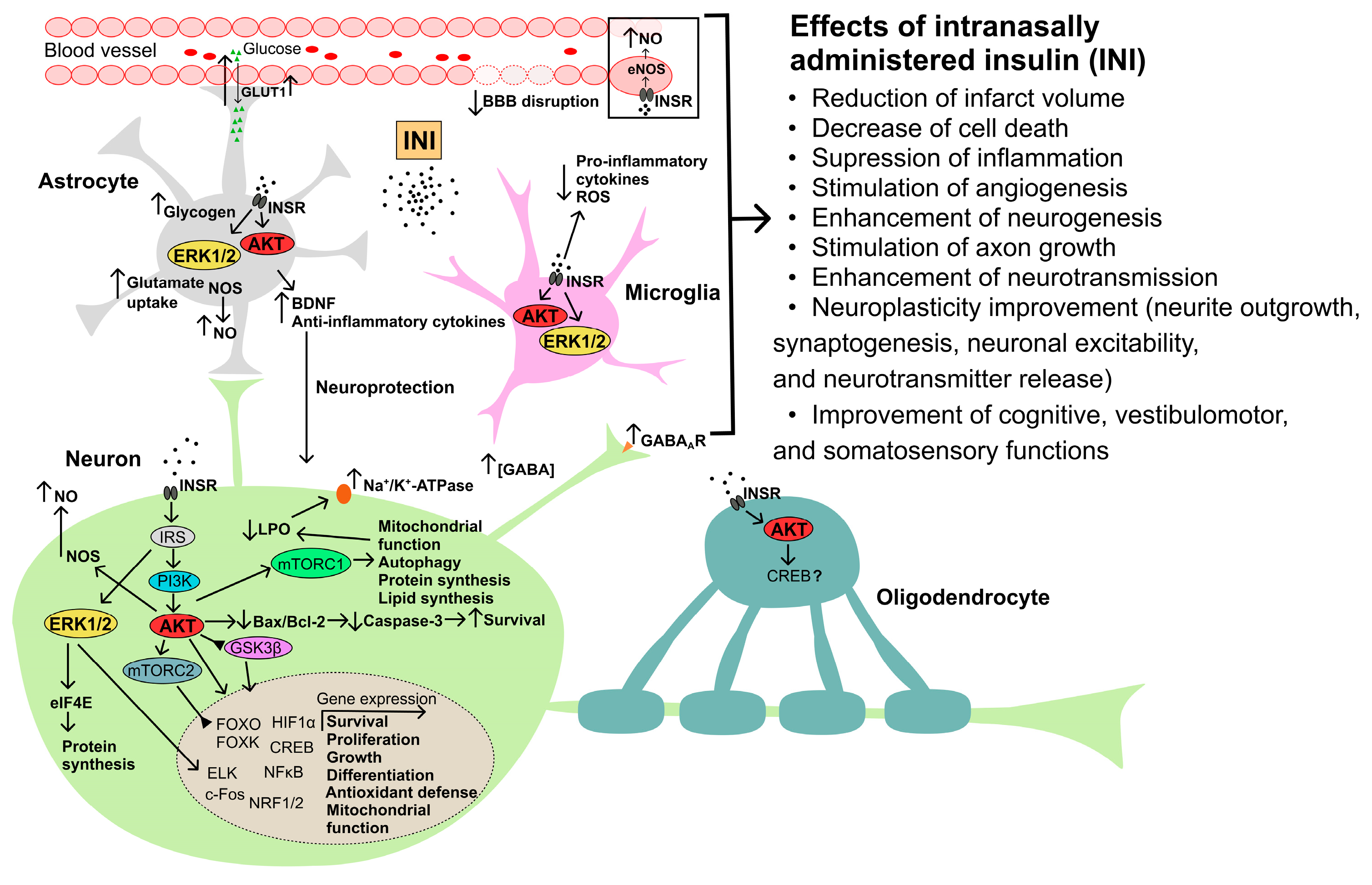
A decrease in the activity of the insulin signaling system of the brain leads to neurodegeneration and impaired regulation of appetite, metabolism, and endocrine functions. This is due to the neuroprotective properties of brain insulin, its leading role in maintaining glucose homeostasis in the brain, as well as in the regulation of the brain signaling network responsible for the functioning of the nervous, endocrine and other systems. One of the approaches to restore the brain insulin system is the use of intranasally administered insulin (INI). INI is being considered as a promising drug to treat Alzheimer's disease and mild cognitive impairment. Clinical application of INI is being developed for the treatment of other neurodegenerative diseases and improve cognitive abilities in stress, overwork, and depression. It has been shown that INI can be used for the treatment of cerebral ischemia, traumatic brain injuries, postoperative delirium, as well as diabetes mellitus and its complications, including dysfunctions in the gonadal and thyroid axes.
1. Introduction
2. Insulin and Insulin Signaling System in the Brain and Intranasal Delivery of Insulin to the Brain
2.1. Sources of Insulin in the Brain
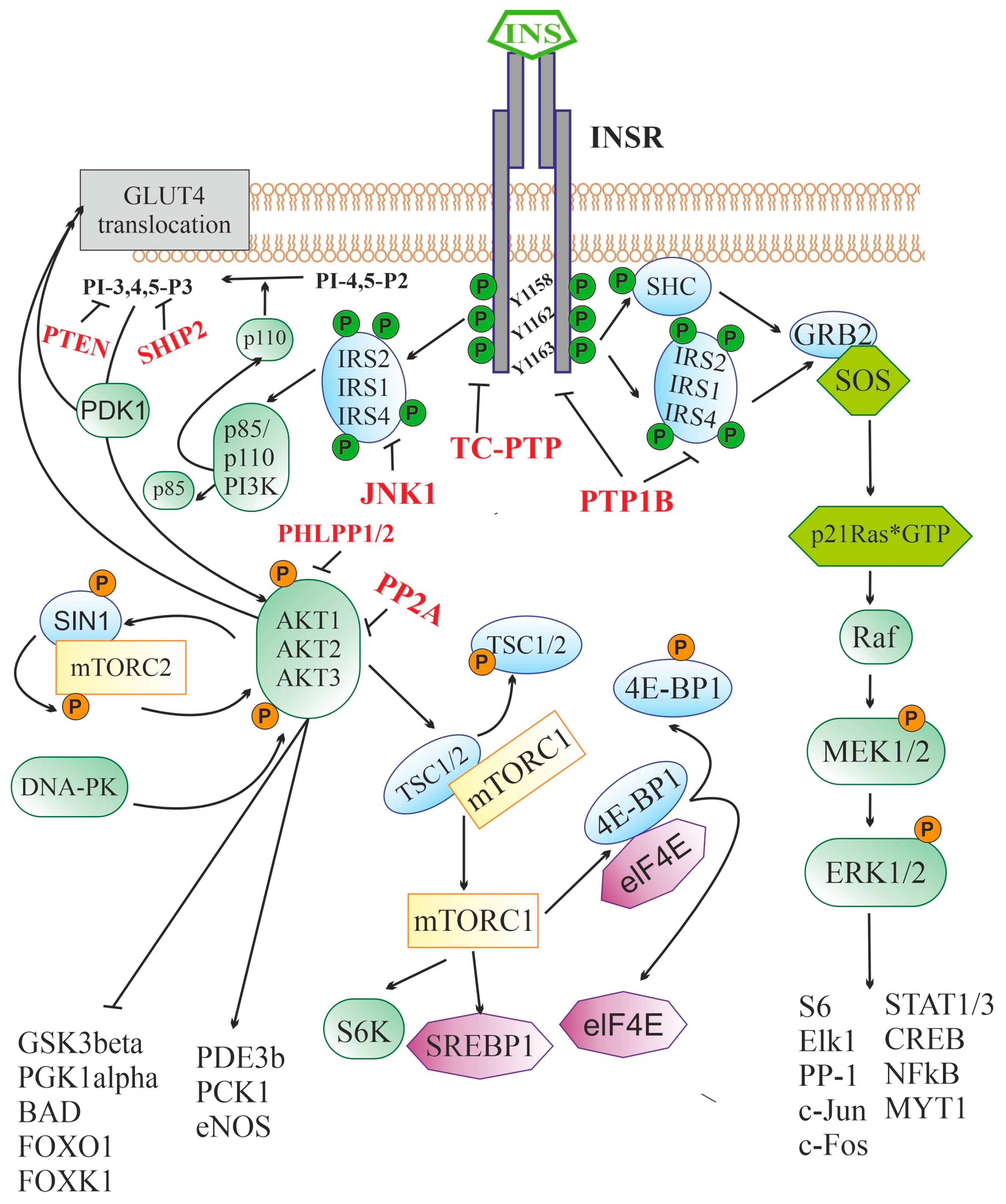
2.2. Insulin Signaling System
2.3. The Intranasal Route of Insulin Delivery
3. Intranasal Insulin and Brain Ischemia
4. Intranasal Insulin and Brain Injury
5. Intranasal Insulin and Diabetes Mellitus
6. Intranasal Insulin and Anesthesia
7. Intranasal Insulin and the Gonadal and Thyroid Systems
References
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829.
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741.
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimer′s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960.
- Cai, Z.; Xiao, M.; Chang, L.; Yan, L.J. Role of insulin resistance in Alzheimer’s disease. Metab. Brain Dis. 2015, 30, 839–851.
- De la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimers Dis. 2018, 62, 1381–1390.
- De la Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83.
- Eraky, S.M.; Ramadan, N.M.; Abo El-Magd, N.F. Ameliorative effects of bromelain on aluminum-induced Alzheimer’s disease in rats through modulation of TXNIP pathway. Int. J. Biol. Macromol. 2022, 227, 1119–1131.
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43.
- Ardanaz, C.G.; Ramírez, M.J.; Solas, M. Brain Metabolic Alterations in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3785.
- González, A.; Calfío, C.; Churruca, M.; Maccioni, R.B. Glucose metabolism and AD: Evidence for a potential diabetes type 3. Alzheimers Res. Ther. 2022, 14, 56.
- Neumann, K.F.; Rojo, L.; Navarrete, L.P.; Farías, G.; Reyes, P.; Maccioni, R.B. Insulin resistance and Alzheimer’s disease: Molecular links & clinical implications. Curr. Alzheimer Res. 2008, 5, 438–447.
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417.
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47.
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814.
- Ortiz, G.G.; Huerta, M.; González-Usigli, H.A.; Torres-Sánchez, E.D.; Delgado-Lara, D.L.; Pacheco-Moisés, F.P.; Mireles-Ramírez, M.A.; Torres-Mendoza, B.M.; Moreno-Cih, R.I.; Velázquez-Brizuela, I.E. Cognitive disorder and dementia in type 2 diabetes mellitus. World J. Diabetes 2022, 13, 319–337.
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52.
- Picone, P.; Sabatino, M.A.; Ditta, L.A.; Amato, A.; San Biagio, P.L.; Mulè, F.; Giacomazza, D.; Dispenza, C.; Di Carlo, M. Nose-to-brain delivery of insulin enhanced by a nanogel carrier. J. Control. Release 2018, 270, 23–36.
- Fan, L.W.; Carter, K.; Bhatt, A.; Pang, Y. Rapid transport of insulin to the brain following intranasal administration in rats. Neural Regen. Res. 2019, 14, 1046–1051.
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci. Rep. 2019, 9, 2621.
- Patel, D.; Patel, B.; Wairkar, S. Intranasal delivery of biotechnology-based therapeutics. Drug Discov. Today 2022, 27, 103371.
- Sharma, M.; Waghela, S.; Mhatre, R.; Saraogi, G.K. A Recent Update on Intranasal Delivery of High Molecular Weight Proteins, Peptides, and Hormones. Curr. Pharm. Des. 2021, 27, 4279–4299.
- Bose, M.; Farias Quipildor, G.; Ehrlich, M.E.; Salton, S.R. Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development. Cells 2022, 11, 3629.
- Duong, V.A.; Nguyen, T.T.; Maeng, H.J. Recent Advances in Intranasal Liposomes for Drug, Gene, and Vaccine Delivery. Pharmaceutics 2023, 15, 207.
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516.
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458.
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38.
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334.
- Novak, P.; Pimentel Maldonado, D.A.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364.
- Novak, V.; Mantzoros, C.S.; Novak, P.; McGlinchey, R.; Dai, W.; Lioutas, V.; Buss, S.; Fortier, C.B.; Khan, F.; Aponte Becerra, L.; et al. MemAID: Memory advancement with intranasal insulin vs. placebo in type 2 diabetes and control participants: A randomized clinical trial. J. Neurol. 2022, 269, 4817–4835.
- Benedict, C.; Frey, W.H., 2nd; Schiöth, H.B.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 2011, 46, 112–115.
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906.
- Ribarič, S. The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules 2016, 21, 689.
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510.
- Chen, J.; Hu, L.; Yang, G.; Hu, Q. Current Therapeutic Strategy in the Nasal Delivery of Insulin: Recent Advances and Future Directions. Curr. Pharm. Biotechnol. 2018, 19, 400–415.
- Muñoz-Jiménez, M.; Zaarkti, A.; García-Arnés, J.A.; García-Casares, N. Antidiabetic Drugs in Alzheimer’s Disease and Mild Cognitive Impairment: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2020, 49, 423–434.
- Gaddam, M.; Singh, A.; Jain, N.; Avanthika, C.; Jhaveri, S.; De la Hoz, I.; Sanka, S.; Goli, S.R. A Comprehensive Review of Intranasal Insulin and Its Effect on the Cognitive Function of Diabetics. Cureus 2021, 13, e17219.
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37.
- Hallschmid, M. Intranasal insulin. J. Neuroendocrinol. 2021, 33, e12934.
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2021, 8, 240–248.
- Miziak, B.; Błaszczyk, B.; Czuczwar, S.J. Some Candidate Drugs for Pharmacotherapy of Alzheimer’s Disease. Pharmaceuticals 2021, 14, 458.
- Long, C.; Han, X.; Yang, Y.; Li, T.; Zhou, Q.; Chen, Q. Efficacy of intranasal insulin in improving cognition in mild cognitive impairment or dementia: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 963933.
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334.
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans: Superiority of insulin aspart. Neuropsychopharmacology 2007, 32, 239–243.
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344.
- Kupila, A.; Sipilä, J.; Keskinen, P.; Simell, T.; Knip, M.; Pulkki, K.; Simell, O. Intranasally administered insulin intended for prevention of type 1 diabetes--a safety study in healthy adults. Diabetes Metab. Res. Rev. 2003, 19, 415–420.
- Schmid, V.; Kullmann, S.; Gfrörer, W.; Hund, V.; Hallschmid, M.; Lipp, H.P.; Häring, H.U.; Preissl, H.; Fritsche, A.; Heni, M. Safety of intranasal human insulin: A review. Diabetes Obes. Metab. 2018, 20, 1563–1577.
- Gwizdala, K.L.; Ferguson, D.P.; Kovan, J.; Novak, V.; Pontifex, M.B. Placebo controlled phase II clinical trial: Safety and efficacy of combining intranasal insulin & acute exercise. Metab. Brain Dis. 2021, 36, 1289–1303.
- Aponte Becerra, L.; Galindo Mendez, B.; Khan, F.; Lioutas, V.; Novak, P.; Mantzoros, C.S.; Ngo, L.H.; Novak, V. Safety of Intranasal Insulin in Type 2 Diabetes on Systemic Insulin: A Double-Blinded Placebo-Controlled Sub-Study of Memaid Trial. Arch. Diabetes Obes. 2022, 4, 403–415.
- Schneider, E.; Spetter, M.S.; Martin, E.; Sapey, E.; Yip, K.P.; Manolopoulos, K.N.; Tahrani, A.A.; Thomas, J.M.; Lee, M.; Hallschmid, M.; et al. The effect of intranasal insulin on appetite and mood in women with and without obesity: An experimental medicine study. Int. J. Obes. 2022, 46, 1319–1327.
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454.
- Schechter, R.; Whitmire, J.; Wheet, G.S.; Beju, D.; Jackson, K.W.; Harlow, R.; Gavin, J.R., III. Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res. 1994, 636, 9–27.
- Gerozissis, K. Brain insulin: Regulation, mechanisms of action and functions. Cell. Mol. Neurobiol. 2003, 23, 1–25.
- Gerozissis, K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur. J. Pharmacol. 2008, 585, 38–49.
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161.
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521.
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487.
- Yu, Y.; Kastin, A.J.; Pan, W. Reciprocal interactions of insulin and insulin-like growth factor I in receptor-mediated transport across the blood-brain barrier. Endocrinology 2006, 147, 2611–2615.
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Effect of diabetes mellitus on the permeability of the blood-brain barrier to insulin. Peptides 1997, 18, 1577–1584.
- Banks, W.A.; Dohgu, S.; Lynch, J.L.; Fleegal-DeMotta, M.A.; Erickson, M.A.; Nakaoke, R.; Vo, T.Q. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology 2008, 149, 1514–1523.
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597.
- May, A.A.; Bedel, N.D.; Shen, L.; Woods, S.C.; Liu, M. Estrogen and insulin transport through the blood-brain barrier. Physiol. Behav. 2016, 163, 312–321.
- May, A.A.; Liu, M.; Woods, S.C.; Begg, D.P. CCK increases the transport of insulin into the brain. Physiol. Behav. 2016, 165, 392–397.
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399.
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314.
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930.
- Beddows, C.A.; Dodd, G.T. Insulin on the brain: The role of central insulin signalling in energy and glucose homeostasis. J. Neuroendocrinol. 2021, 33, e12947.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561.
- Villaseñor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Grüninger, F.; Loetscher, H.; Freskgård, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. J. Cereb. Blood Flow Metab. 2017, 37, 3683–3694.
- Mäe, M.A.; He, L.; Nordling, S.; Vazquez-Liebanas, E.; Nahar, K.; Jung, B.; Li, X.; Tan, B.C.; Chin Foo, J.; Cazenave-Gassiot, A.; et al. Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circ. Res. 2021, 128, e46–e62.
- McKinley, M.J.; Denton, D.A.; Ryan, P.J.; Yao, S.T.; Stefanidis, A.; Oldfield, B.J. From sensory circumventricular organs to cerebral cortex: Neural pathways controlling thirst and hunger. J. Neuroendocrinol. 2019, 31, e12689.
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962.
- Porniece Kumar, M.; Cremer, A.L.; Klemm, P.; Steuernagel, L.; Sundaram, S.; Jais, A.; Hausen, A.C.; Tao, J.; Secher, A.; Pedersen, T.Å.; et al. Insulin signalling in tanycytes gates hypothalamic insulin uptake and regulation of AgRP neuron activity. Nat. Metab. 2021, 3, 1662–1679.
- Langlet, F.; Levin, B.E.; Luquet, S.; Mazzone, M.; Messina, A.; Dunn-Meynell, A.A.; Balland, E.; Lacombe, A.; Mazur, D.; Carmeliet, P.; et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell. Metab. 2013, 17, 607–617.
- Prevot, V.; Langlet, F.; Dehouck, B. Flipping the tanycyte switch: How circulating signals gain direct access to the metabolic brain. Aging 2013, 5, 332–334.
- Morita-Takemura, S.; Wanaka, A. Blood-to-brain communication in the hypothalamus for energy intake regulation. Neurochem. Int. 2019, 28, 135–142.
- Dorn, A.; Bernstein, H.G.; Hahn, H.J.; Ziegler, M.; Rummelfanger, H. Insulin immunohistochemistry of rodent CNS: Apparent species differences but good correlation with radioimmunological data. Histochemistry 1981, 71, 609–616.
- Dorn, A.; Bernstein, H.G.; Rinne, A.; Hahn, H.J.; Ziegler, M. Insulin-like immunoreactivity in the human brain—A preliminary report. Histochemistry 1982, 74, 293–300.
- Baskin, D.G.; Brewitt, B.; Davidson, D.A.; Corp, E.; Paquette, T.; Figlewicz, D.P.; Lewellen, T.K.; Graham, M.K.; Woods, S.G.; Dorsa, D.M. Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes 1986, 35, 246–249.
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic localization of insulin receptors in rat brain: Prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138.
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570.
- Unger, J.; McNeill, T.H.; Moxley, R.T., 3rd; White, M.; Moss, A.; Livingston, J.N. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 1989, 31, 143–157.
- Marks, J.L.; Porte, D., Jr.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236.
- Iozzo, P.; Osman, S.; Glaser, M.; Knickmeier, M.; Ferrannini, E.; Pike, V.W.; Camici, P.G.; Law, M.P. In vivo imaging of insulin receptors by PET: Preclinical evaluation of iodine-125 and iodine-124 labelled human insulin. Nucl. Med. Biol. 2002, 29, 73–82.
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81.
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin action on astrocytes: From energy homeostasis to behaviour. J. Neuroendocrinol. 2021, 33, e12953.
- Moller, D.E.; Yokota, A.; Caro, J.F.; Flier, J.S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 1989, 3, 1263–1269.
- Seino, S.; Bell, G.I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 1989, 159, 312–316.
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413.
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118.
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431.
- Escribano, O.; Beneit, N.; Rubio-Longás, C.; López-Pastor, A.R.; Gómez-Hernández, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206.
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221.
- Drakas, R.; Tu, X.; Baserga, R. Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 9272–9276.
- De Meyts, P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 2008, 33, 376–384.
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623.
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304.
- Razzini, G.; Ingrosso, A.; Brancaccio, A.; Sciacchitano, S.; Esposito, D.L.; Falasca, M. Different subcellular localization and phosphoinositides binding of insulin receptor substrate protein pleckstrin homology domains. Mol. Endocrinol. 2000, 14, 823–836.
- Sadagurski, M.; Dong, X.C.; Myers, M.G., Jr.; White, M.F. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2013, 3, 55–63.
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243.
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582.
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341.
- Levenga, J.; Wong, H.; Milstead, R.A.; Keller, B.N.; LaPlante, L.E.; Hoeffer, C.A. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. eLife 2017, 6, e30640.
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405.
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189.
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911.
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776.
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131.
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192.
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimers Dis. 2011, 2011, 189728.
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848.
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105.
- Kim, K.W.; Donato, J., Jr.; Berglund, E.D.; Choi, Y.H.; Kohno, D.; Elias, C.F.; Depinho, R.A.; Elmquist, J.K. FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Invest. 2012, 122, 2578–2589.
- Ren, H.; Plum-Morschel, L.; Gutierrez-Juarez, R.; Lu, T.Y.; Kim-Muller, J.Y.; Heinrich, G.; Wardlaw, S.L.; Silver, R.; Accili, D. Blunted refeeding response and increased locomotor activity in mice lacking FoxO1 in synapsin-Cre-expressing neurons. Diabetes 2013, 62, 3373–3383.
- Du, S.; Zheng, H. Role of FoxO transcription factors in aging and age-related metabolic and neurodegenerative diseases. Cell Biosci. 2021, 11, 188.
- Oli, V.; Gupta, R.; Kumar, P. FOXO and related transcription factors binding elements in the regulation of neurodegenerative disorders. J. Chem. Neuroanat. 2021, 116, 102012.
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171.
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology 2006, 21, 362–369.
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796.
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234.
- Bakke, J.; Haj, F.G. Protein-tyrosine phosphatase 1B substrates and metabolic regulation. Semin. Cell Dev. Biol. 2015, 37, 58–65.
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674.
- Dodd, G.T.; Xirouchaki, C.E.; Eramo, M.; Mitchell, C.A.; Andrews, Z.B.; Henry, B.A.; Cowley, M.A.; Tiganis, T. Intranasal Targeting of Hypothalamic PTP1B and TCPTP Reinstates Leptin and Insulin Sensitivity and Promotes Weight Loss in Obesity. Cell Rep. 2019, 28, 2905–2922.e5.
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537.
- Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317.
- Nogueiras, R.; Sabio, G. Brain JNK and metabolic disease. Diabetologia 2021, 64, 265–274.
- Busquets, O.; Espinosa-Jiménez, T.; Ettcheto, M.; Olloquequi, J.; Bulló, M.; Carro, E.; Cantero, J.L.; Casadesús, G.; Folch, J.; Verdaguer, E.; et al. JNK1 and JNK3: Divergent functions in hippocampal metabolic-cognitive function. Mol. Med. 2022, 28, 48.
- Accardi, G.; Virruso, C.; Balistreri, C.R.; Emanuele, F.; Licastro, F.; Monastero, R.; Porcellini, E.; Vasto, S.; Verga, S.; Caruso, C.; et al. SHIP2: A "new" insulin pathway target for aging research. Rejuvenation Res. 2014, 17, 221–225.
- Sekar, S.; Taghibiglou, C. Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci. Lett. 2018, 666, 139–143.
- Zhao, J.; Yin, L.; Jiang, L.; Hou, L.; He, L.; Zhang, C. PTEN nuclear translocation enhances neuronal injury after hypoxia-ischemia via modulation of the nuclear factor-κB signaling pathway. Aging 2021, 13, 16165–16177.
- Javadpour, P.; Dargahi, L.; Ahmadiani, A.; Ghasemi, R. To be or not to be: PP2A as a dual player in CNS functions, its role in neurodegeneration, and its interaction with brain insulin signaling. Cell. Mol. Life Sci. 2019, 76, 2277–2297.
- Frey, W.H., II. Neurologic Agents for Nasal Administration to the Brain. PCT International Patent WO 91/07947, 13 June 1991.
- Frey, W.H., II. Method of Administering Neurologic Agents to the Brain. U.S. Patent 5,624,898, 29 April 1997.
- Frey, W.H., II. Method for Administering Insulin to the Brain. U.S. Patent 6,313,093 B1, 6 November 2001.
- Nedelcovych, M.T.; Gadiano, A.J.; Wu, Y.; Manning, A.A.; Thomas, A.G.; Khuder, S.S.; Yoo, S.W.; Xu, J.; McArthur, J.C.; Haughey, N.J.; et al. Pharmacokinetics of Intranasal versus Subcutaneous Insulin in the Mouse. ACS Chem. Neurosci. 2018, 9, 809–816.
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309.
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628.
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H., II. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496.
- Salameh, T.S.; Bullock, K.M.; Hujoel, I.A.; Niehoff, M.L.; Wolden-Hanson, T.; Kim, J.; Morley, J.E.; Farr, S.A.; Banks, W.A. Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J. Alzheimers Dis. 2015, 47, 715–728.
- Chudoba, C.; Kleinridders, A. 271-LB: Intranasal Insulin Treatment Causes Sex-Specific Differences in Metabolism and Behavior in Mice. Diabetes 2019, 68 (Suppl. 1), 271-LB.
- Jauch-Chara, K.; Friedrich, A.; Rezmer, M.; Melchert, U.H.; Scholand-Engler, H.G.; Hallschmid, M.; Oltmanns, K.M. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes 2012, 61, 2261–2268.
- Leary, A.C.; Stote, R.M.; Breedt, H.J.; O’Brien, J.; Buckley, B. Pharmacokinetics and pharmacodynamics of intranasal insulin administered to healthy subjects in escalating doses. Diabetes Technol. Ther. 2005, 7, 124–130.
- Leker, R.R.; Shohami, E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: Neuroprotective opportunities. Brain Res. Brain Res. Rev. 2002, 39, 55–73.
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G.; et al. Stroke Thrombolysis Trialists’ Collaborative Group. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: A meta-analysis of individual patient data from randomised trials. Lancet 2014, 384, 1929–1935.
- Zhou, Z.B.; Meng, L.; Gelb, A.W.; Lee, R.; Huang, W.Q. Cerebral ischemia during surgery: An overview. J. Biomed. Res. 2016, 30, 83–87.
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215.
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J. Neuroinflamm. 2010, 7, 14.
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966.
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107.
- Dharmasaroja, P.A. Fluid Intake Related to Brain Edema in Acute Middle Cerebral Artery Infarction. Transl. Stroke Res. 2016, 7, 49–53.
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients with Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418.
- Greenberg, S.M.; Ziai, W.C.; Cordonnier, C.; Dowlatshahi, D.; Francis, B.; Goldstein, J.N.; Hemphill, J.C., 3rd; Johnson, R.; Keigher, K.M.; Mack, W.J.; et al. American Heart Association/American Stroke Association. 2022 Guideline for the Management of Patients with Spontaneous Intracerebral Hemorrhage: A Guideline from the American Heart Association/American Stroke Association. Stroke 2022, 53, e282–e361.
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432.
- Zhang, Z.; Yan, J.; Shi, H. Hyperglycemia as a Risk Factor of Ischemic Stroke. J. Drug Metab. Toxicol. 2013, 4, 153.
- Palaiodimou, L.; Lioutas, V.A.; Lambadiari, V.; Paraskevas, G.P.; Voumvourakis, K.; Tsivgoulis, G. Glycemia management in acute ischemic stroke: Current concepts and novel therapeutic targets. Postgrad. Med. 2019, 131, 423–437.
- Middleton, S.; McElduff, P.; Ward, J.; Grimshaw, J.M.; Dale, S.; D’Este, C.; Drury, P.; Griffiths, R.; Cheung, N.W.; Quinn, C.; et al. QASC Trialists Group. Implementation of evidence-based treatment protocols to manage fever, hyperglycaemia, and swallowing dysfunction in acute stroke (QASC): A cluster randomised controlled trial. Lancet 2011, 378, 1699–1706.
- Passero, S.; Ciacci, G.; Ulivelli, M. The influence of diabetes and hyperglycemia on clinical course after intracerebral hemorrhage. Neurology 2003, 61, 1351–1356.
- Kim, Y.; Han, M.H.; Kim, C.H.; Kim, J.M.; Cheong, J.H.; Ryu, J.I. Increased Short-Term Mortality in Patients with Spontaneous Intracerebral Hemorrhage and its Association with Admission Glucose Levels and Leukocytosis. World Neurosurg. 2017, 98, 503–511.
- Almdal, T.; Scharling, H.; Jensen, J.S.; Vestergaard, H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: A population-based study of 13,000 men and women with 20 years of follow-up. Arch. Intern. Med. 2004, 164, 1422–1426.
- Banerjee, C.; Moon, Y.P.; Paik, M.C.; Rundek, T.; Mora-McLaughlin, C.; Vieira, J.R.; Sacco, R.L.; Elkind, M.S. Duration of diabetes and risk of ischemic stroke: The Northern Manhattan Study. Stroke 2012, 43, 1212–1217.
- Mitsios, J.P.; Ekinci, E.I.; Mitsios, G.P.; Churilov, L.; Thijs, V. Relationship Between Glycated Hemoglobin and Stroke Risk: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e007858.
- Pan, Y.; Chen, W.; Wang, Y. Prediabetes and Outcome of Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Meta-analysis. J. Stroke Cerebrovasc. Dis. 2019, 28, 683–692.
- Kernan, W.N.; Inzucchi, S.E.; Viscoli, C.M.; Brass, L.M.; Bravata, D.M.; Shulman, G.I.; McVeety, J.C.; Horwitz, R.I. Impaired insulin sensitivity among nondiabetic patients with a recent TIA or ischemic stroke. Neurology 2003, 60, 1447–1451.
- Hishinuma, A.; Majima, M.; Kurabayashi, H. Insulin resistance in patients with stroke is related to visceral fat obesity and adipocytokines. J. Stroke Cerebrovasc. Dis. 2008, 17, 175–180.
- Voll, C.L.; Auer, R.N. The effect of postischemic blood glucose levels on ischemic brain damage in the rat. Ann. Neurol. 1988, 24, 638–646.
- Voll, C.L.; Auer, R.N. Insulin attenuates ischemic brain damage independent of its hypoglycemic effect. J. Cereb. Blood Flow Metab. 1991, 11, 1006–1014.
- Voll, C.L.; Whishaw, I.Q.; Auer, R.N. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke 1989, 20, 646–651.
- Hamilton, M.G.; Tranmer, B.I.; Auer, R.N. Insulin reduction of cerebral infarction due to transient focal ischemia. J. Neurosurg. 1995, 82, 262–268.
- Guyot, L.L.; Diaz, F.G.; O’Regan, M.H.; Ren, J.; Phillis, J.W. The effect of intravenous insulin on accumulation of excitotoxic and other amino acids in the ischemic rat cerebral cortex. Neurosci. Lett. 2000, 288, 61–65.
- Fukuoka, S.; Yeh, H.; Mandybur, T.I.; Tew, J.M., Jr. Effect of insulin on acute experimental cerebral ischemia in gerbils. Stroke 1989, 20, 396–399.
- Meden, P.; Andersen, M.; Overgaard, K.; Rasmussen, R.S.; Boysen, G. The effects of early insulin treatment combined with thrombolysis in rat embolic stroke. Neurol. Res. 2002, 24, 399–404.
- Huang, S.S.; Lu, Y.J.; Huang, J.P.; Wu, Y.T.; Day, Y.J.; Hung, L.M. The essential role of endothelial nitric oxide synthase activation in insulin-mediated neuroprotection against ischemic stroke in diabetes. J. Vasc. Surg. 2014, 59, 483–491.
- Izumi, Y.; Pinard, E.; Roussel, S.; Seylaz, J. Insulin protects brain tissue against focal ischemia in rats. Neurosci. Lett. 1992, 144, 121–123.
- Fanne, R.A.; Nassar, T.; Heyman, S.N.; Hijazi, N.; Higazi, A.A. Insulin and glucagon share the same mechanism of neuroprotection in diabetic rats: Role of glutamate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R668–R673.
- Guan, J.; Williams, C.; Gunning, M.; Mallard, C.; Gluckman, P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J. Cereb. Blood Flow Metab. 1993, 13, 609–616.
- Zhu, C.Z.; Auer, R.N. Intraventricular administration of insulin and IGF-1 in transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 237–242.
- Russo, V.; Candeloro, P.; Malara, N.; Perozziello, G.; Iannone, M.; Scicchitano, M.; Mollace, R.; Musolino, V.; Gliozzi, M.; Carresi, C.; et al. Key Role of Cytochrome C for Apoptosis Detection Using Raman Microimaging in an Animal Model of Brain Ischemia with Insulin Treatment. Appl. Spectrosc. 2019, 73, 1208–1217.
- Sanderson, T.H.; Kumar, R.; Sullivan, J.M.; Krause, G.S. Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-XL binding. J. Neurochem. 2008, 106, 1248–1258.
- Sanderson, T.H.; Kumar, R.; Murariu-Dobrin, A.C.; Page, A.B.; Krause, G.S.; Sullivan, J.M. Insulin activates the PI3K-Akt survival pathway in vulnerable neurons following global brain ischemia. Neurol. Res. 2009, 31, 947–958.
- Duarte, A.I.; Santos, P.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 2008, 1783, 994–1002.
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017.
- McKay, M.K.; Hester, R.L. Role of nitric oxide, adenosine, and ATP-sensitive potassium channels in insulin-induced vasodilation. Hypertension 1996, 28, 202–208.
- Hung, L.M.; Huang, J.P.; Liao, J.M.; Yang, M.H.; Li, D.E.; Day, Y.J.; Huang, S.S. Insulin renders diabetic rats resistant to acute ischemic stroke by arresting nitric oxide reaction with superoxide to form peroxynitrite. J. Biomed. Sci. 2014, 21, 92.
- Hughes, T.M.; Craft, S. The role of insulin in the vascular contributions to age-related dementia. Biochim. Biophys. Acta 2016, 1862, 983–991.
- Muniyappa, R.; Yavuz, S. Metabolic actions of angiotensin II and insulin: A microvascular endothelial balancing act. Mol. Cell. Endocrinol. 2013, 378, 59–69.
- Akintola, A.A.; van Opstal, A.M.; Westendorp, R.G.; Postmus, I.; van der Grond, J.; van Heemst, D. Effect of intranasally administered insulin on cerebral blood flow and perfusion; a randomized experiment in young and older adults. Aging 2017, 9, 790–802.
- Lioutas, V.A.; Alfaro-Martinez, F.; Bedoya, F.; Chung, C.C.; Pimentel, D.A.; Novak, V. Intranasal Insulin and Insulin-Like Growth Factor 1 as Neuroprotectants in Acute Ischemic Stroke. Transl. Stroke Res. 2015, 6, 264–275.
- Zorina, I.I.; Zakharova, I.O.; Bayunova, L.V.; Avrova, N.F. Insulin Administration Prevents Accumulation of Conjugated Dienes and Trienes and Inactivation of Na+, K+-ATPase in the Rat Cerebral Cortex during Two-Vessel Forebrain Ischemia and Reperfusion. J. Evol. Biochem. Physiol. 2018, 54, 246–249.
- Zorina, I.I.; Galkina, O.V.; Bayunova, L.V.; Zakharova, I.O. Effect of Insulin on Lipid Peroxidation and Glutathione Levels in a Two-Vessel Occlusion Model of Rat Forebrain Ischemia Followed by Reperfusion. J. Evol. Biochem. Physiol. 2019, 55, 333–335.
- Zakharova, I.O.; Bayunova, L.V.; Zorina, I.I.; Sokolova, T.V.; Shpakov, A.O.; Avrova., N.F. Insulin and α-Tocopherol Enhance the Protective Effect of Each Other on Brain Cortical Neurons under Oxidative Stress Conditions and in Rat Two-Vessel Forebrain Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 11768.
- Zakharova, I.O.; Bayunova, L.V.; Zorina, I.I.; Shpakov, A.O.; Avrova, N.F. Insulin and Brain Gangliosides Prevent Metabolic Disorders Caused by Activation of Free Radical Reactions after Two-Vessel Ischemia–Reperfusion Injury to the Rat Forebrain. J. Evol. Biochem. Physiol. 2022, 58, 279–291.
- Xu, L.B.; Huang, H.D.; Zhao, M.; Zhu, G.C.; Xu, Z. Intranasal Insulin Treatment Attenuates Metabolic Distress and Early Brain Injury After Subarachnoid Hemorrhage in Mice. Neurocrit. Care 2021, 34, 154–166.
- Zhu, Y.; Huang, Y.; Yang, J.; Tu, R.; Zhang, X.; He, W.W.; Hou, C.Y.; Wang, X.M.; Yu, J.M.; Jiang, G.H. Intranasal insulin ameliorates neurological impairment after intracerebral hemorrhage in mice. Neural Regen. Res. 2022, 17, 210–216.
- Liu, X.F.; Fawcett, J.R.; Thorne, R.G.; DeFor, T.A.; Frey, W.H., II. Intranasal administration of insulin-like growth factor-I bypasses the blood-brain barrier and protects against focal cerebral ischemic damage. J. Neurol. Sci. 2001, 187, 91–97.
- Liu, X.F.; Fawcett, J.R.; Thorne, R.G.; Frey, W.H., II. Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci. Lett. 2001, 308, 91–94.
- Liu, X.F.; Fawcett, J.R.; Hanson, L.R.; Frey, W.H., II. The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin-like growth factor-I in rats. J. Stroke Cerebrovasc. Dis. 2004, 13, 16–23.
- Fletcher, L.; Kohli, S.; Sprague, S.M.; Scranton, R.A.; Lipton, S.A.; Parra, A.; Jimenez, D.F.; Digicaylioglu, M. Intranasal delivery of erythropoietin plus insulin-like growth factor-I for acute neuroprotection in stroke. Laboratory investigation. J. Neurosurg. 2009, 111, 164–170.
- Cai, Z.; Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G. Intranasal administration of insulin-like growth factor-1 protects against lipopolysaccharide-induced injury in the developing rat brain. Neuroscience 2011, 194, 195–207.
- Lin, S.; Rhodes, P.G.; Cai, Z. Whole body hypothermia broadens the therapeutic window of intranasally administered IGF-1 in a neonatal rat model of cerebral hypoxia-ischemia. Brain Res. 2011, 1385, 246–256.
- Shen, H.; Gu, X.; Wei, Z.Z.; Wu, A.; Liu, X.; Wei, L. Combinatorial intranasal delivery of bone marrow mesenchymal stem cells and insulin-like growth factor-1 improves neurovascularization and functional outcomes following focal cerebral ischemia in mice. Exp. Neurol. 2021, 337, 113542.
- Iravanpour, F.; Dargahi, L.; Rezaei, M.; Haghani, M.; Heidari, R.; Valian, N.; Ahmadiani, A. Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson’s disease. CNS Neurosci. Ther. 2021, 27, 308–319.
- Kim, B.H.; Kelschenbach, J.; Borjabad, A.; Hadas, E.; He, H.; Potash, M.J.; Nedelcovych, M.T.; Rais, R.; Haughey, N.J.; McArthur, J.C.; et al. Intranasal insulin therapy reverses hippocampal dendritic injury and cognitive impairment in a model of HIV-associated neurocognitive disorders in EcoHIV-infected mice. AIDS 2019, 33, 973–984.
- Rassovsky, Y.; Levi, Y.; Agranov, E.; Sela-Kaufman, M.; Sverdlik, A.; Vakil, E. Predicting long-term outcome following traumatic brain injury (TBI). J. Clin. Exp. Neuropsychol. 2015, 37, 354–366.
- Røe, C.; Sveen, U.; Alvsåker, K.; Bautz-Holter, E. Post-concussion symptoms after mild traumatic brain injury: Influence of demographic factors and injury severity in a 1-year cohort study. Disabil. Rehabil. 2009, 31, 1235–1243.
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75, S24–S33.
- Gross, H.; Kling, A.; Henry, G.; Herndon, C.; Lavretsky, H. Local cerebral glucose metabolism in patients with long-term behavioral and cognitive deficits following mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 1996, 8, 324–334.
- Toth, A. Magnetic Resonance Imaging Application in the Area of Mild and Acute Traumatic Brain Injury: Implications for Diagnostic Markers? In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2015; Chapter 24; ISBN 978-1-4665-6598-2.
- Llompart-Pou, J.A.; Raurich, J.M.; Pérez-Bárcena, J.; Barceló, A.; Ibáñez, J.; Ayestarán, J.I. Acute Hypothalamic-pituitary-adrenal response in traumatic brain injury with and without extracerebral trauma. Neurocrit. Care 2008, 9, 230–236.
- Shaughness, M.; Acs, D.; Brabazon, F.; Hockenbury, N.; Byrnes, K.R. Role of Insulin in Neurotrauma and Neurodegeneration: A Review. Front. Neurosci. 2020, 14, 547175.
- Arora, P.; Singh, K.; Kumari, M.; Trivedi, R. Temporal profile of serum metabolites and inflammation following closed head injury in rats is associated with HPA axis hyperactivity. Metabolomics 2022, 18, 28.
- Karelina, K.; Sarac, B.; Freeman, L.M.; Gaier, K.R.; Weil, Z.M. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 2016, 43, 1034–1043.
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228.
- Selwyn, R.; Hockenbury, N.; Jaiswal, S.; Mathur, S.; Armstrong, R.C.; Byrnes, K.R. Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J. Neurotrauma 2013, 30, 1943–1953.
- Yoshino, A.; Hovda, D.A.; Kawamata, T.; Katayama, Y.; Becker, D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991, 561, 106–119.
- Brabazon, F.; Wilson, C.M.; Shukla, D.K.; Mathur, S.; Jaiswal, S.; Bermudez, S.; Byrnes, K.R.; Selwyn, R. FDG-PET Combined with MRI Elucidates the Pathophysiology of Traumatic Brain Injury in Rats. J. Neurotrauma 2017, 34, 1074–1085.
- Yasmin, A.; Jokivarsi, K.; Poutiainen, P.; Pitkänen, A.; Gröhn, O.; Immonen, R. Chronic hypometabolism in striatum and hippocampal network after traumatic brain injury and their relation with memory impairment—-FDG-PET and MRI 4 months after fluid percussion injury in rat. Brain Res. 2022, 1788, 147934.
- Brabazon, F.; Wilson, C.M.; Jaiswal, S.; Reed, J.; Frey, W.H., 2nd; Byrnes, K.R. Intranasal insulin treatment of an experimental model of moderate traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 3203–3218.
- Zhang, J.; Zhang, Y.; Zou, J.; Cao, F. A meta-analysis of cohort studies: Traumatic brain injury and risk of Alzheimer’s Disease. PLoS ONE 2021, 16, e0253206.
- Colca, J.R.; Finck, B.N. Metabolic Mechanisms Connecting Alzheimer’s and Parkinson’s Diseases: Potential Avenues for Novel Therapeutic Approaches. Front. Mol. Biosci. 2022, 9, 929328.
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cereb. Blood Flow Metab. 2004, 24, 133–150.
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Prim. 2018, 4, 18003.
- Nijssen, K.M.R.; Mensink, R.P.; Joris, P.J. Effects of Intranasal Insulin Administration on Cerebral Blood Flow and Cognitive Performance in Adults: A Systematic Review of Randomized, Placebo-Controlled Intervention Studies. Neuroendocrinology 2022, 24, 1–13.
- Blázquez, E.; Hurtado-Carneiro, V.; LeBaut-Ayuso, Y.; Velázquez, E.; García-García, L.; Gómez-Oliver, F.; Ruiz-Albusac, J.M.; Ávila, J.; Pozo, M.Á. Significance of Brain Glucose Hypometabolism, Altered Insulin Signal Transduction, and Insulin Resistance in Several Neurological Diseases. Front. Endocrinol. 2022, 13, 873301.
- Lee, T.H.; Yau, S.Y. From Obesity to Hippocampal Neurodegeneration: Pathogenesis and Non-Pharmacological Interventions. Int. J. Mol. Sci. 2020, 22, 201.
- Sharma, S. High fat diet and its effects on cognitive health: Alterations of neuronal and vascular components of brain. Physiol. Behav. 2021, 240, 113528.
- Al Haj Ahmad, R.M.; Ababneh, N.A.; Al-Domi, H.A. Brain insulin resistance as a mechanistic mediator links peripheral metabolic disorders with declining cognition. Diabetes Metab. Syndr. 2022, 16, 102468.
- Dutta, B.J.; Singh, S.; Seksaria, S.; Das Gupta, G.; Singh, A. Inside the diabetic brain: Insulin resistance and molecular mechanism associated with cognitive impairment and its possible therapeutic strategies. Pharmacol. Res. 2022, 182, 106358.
- Zhang, Q.; Jin, K.; Chen, B.; Liu, R.; Cheng, S.; Zhang, Y.; Lu, J. Overnutrition Induced Cognitive Impairment: Insulin Resistance, Gut-Brain Axis, and Neuroinflammation. Front. Neurosci. 2022, 16, 884579.
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181.
- Schulingkamp, R.J.; Pagano, T.C.; Hung, D.; Raffa, R.B. Insulin receptors and insulin action in the brain: Review and clinical implications. Neurosci. Biobehav. Rev. 2000, 24, 855–872.
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711.
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Häring, H.U.; Fritsche, A.; Preissl, H. Selective insulin resistance in homeostatic and cognitive control brain areas in overweight and obese adults. Diabetes Care 2015, 38, 1044–1050.
- Brands, A.M.; Kessels, R.P.; de Haan, E.H.; Kappelle, L.J.; Biessels, G.J. Cerebral dysfunction in type 1 diabetes: Effects of insulin, vascular risk factors and blood-glucose levels. Eur. J. Pharmacol. 2004, 490, 159–168.
- Wicha, P.; Das, S.; Mahakkanukrauh, P. Blood-brain barrier dysfunction in ischemic stroke and diabetes: The underlying link, mechanisms and future possible therapeutic targets. Anat. Cell Biol. 2021, 54, 165–177.
- Romanova, I.V.; Derkach, K.V.; Mikhrina, A.L.; Sukhov, I.B.; Mikhailova, E.V.; Shpakov, A.O. The Leptin, Dopamine and Serotonin Receptors in Hypothalamic POMC-Neurons of Normal and Obese Rodents. Neurochem. Res. 2018, 43, 821–837.
- Derkach, K.; Zakharova, I.; Zorina, I.; Bakhtyukov, A.; Romanova, I.; Bayunova, L.; Shpakov, A. The evidence of metabolic-improving effect of metformin in Ay/a mice with genetically-induced melanocortin obesity and the contribution of hypothalamic mechanisms to this effect. PLoS ONE 2019, 14, e0213779.
- Schuh, A.F.; Rieder, C.M.; Rizzi, L.; Chaves, M.; Roriz-Cruz, M. Mechanisms of brain aging regulation by insulin: Implications for neurodegeneration in late-onset Alzheimer’s disease. ISRN Neurol. 2011, 2011, 306905.
- Matioli, M.N.P.S.; Nitrini, R. Mechanisms linking brain insulin resistance to Alzheimer’s disease. Dement. Neuropsychol. 2015, 9, 96–102.
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357.
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvourakis, K.I.; et al. Type 2 Diabetes Mellitus as a Risk Factor for Alzheimer’s Disease: Review and Meta-Analysis. Biomedicines 2022, 10, 778.
- Santiago, J.C.P.; Hallschmid, M. Outcomes and clinical implications of intranasal insulin administration to the central nervous system. Exp. Neurol. 2019, 317, 180–190.
- Riederer, P.; Korczyn, A.D.; Ali, S.S.; Bajenaru, O.; Choi, M.S.; Chopp, M.; Dermanovic-Dobrota, V.; Grünblatt, E.; Jellinger, K.A.; Kamal, M.A.; et al. The diabetic brain and cognition. J. Neural Transm. 2017, 124, 1431–1454.
- Li, T.; Cao, H.X.; Ke, D. Type 2 Diabetes Mellitus Easily Develops into Alzheimer’s Disease via Hyperglycemia and Insulin Resistance. Curr. Med. Sci. 2021, 41, 1165–1171.
- Novak, V.; Milberg, W.; Hao, Y.; Munshi, M.; Novak, P.; Galica, A.; Manor, B.; Roberson, P.; Craft, S.; Abduljalil, A. Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care 2014, 37, 751–759.
- Zhang, H.; Hao, Y.; Manor, B.; Novak, P.; Milberg, W.; Zhang, J.; Fang, J.; Novak, V. Intranasal insulin enhanced resting-state functional connectivity of hippocampal regions in type 2 diabetes. Diabetes 2015, 64, 1025–1034.
- Wang, Y.; Sun, L.; He, G.; Gang, X.; Zhao, X.; Wang, G.; Ning, G. Cerebral perfusion alterations in type 2 diabetes mellitus—A systematic review. Front. Neuroendocrinol. 2021, 62, 100916.
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166.
- Caplan, G.A.; Lan, Z.; Newton, L.; Kvelde, T.; McVeigh, C.; Hill, M.A. Transcranial Doppler to measure cerebral blood flow in delirium superimposed on dementia. A cohort study. J. Am. Med. Dir. Assoc. 2014, 15, 355–360.
- Maldonado, J.R. Delirium pathophysiology: An updated hypothesis of the etiology of acute brain failure. Int. J. Geriatr. Psychiatry 2018, 33, 1428–1457.
- Caplan, G.A.; Kvelde, T.; Lai, C.; Yap, S.L.; Lin, C.; Hill, M.A. Cerebrospinal fluid in long-lasting delirium compared with Alzheimer’s dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1130–1136.
- Haggstrom, L.R.; Nelson, J.A.; Wegner, E.A.; Caplan, G.A. 2-18F-fluoro-2-deoxyglucose positron emission tomography in delirium. J. Cereb. Blood Flow Metab. 2017, 37, 3556–3567.
- Ma, H.; Liao, Y.; Mo, Y.; Li, Z.-R.; Liao, Q.; Wang, Y.-C.; Duan, K.-M.; Chen, M.-H.; Ouyang, W. Decreased cerebral glucose metabolism in elderly patients with postoperative delirium: A case-control study. J. Anesth. Perioper. Med. 2017, 4, 162–168.
- Evered, L.; Silbert, B.; Knopman, D.S.; Scott, D.A.; DeKosky, S.T.; Rasmussen, L.S.; Oh, E.S.; Crosby, G.; Berger, M.; Eckenhoff, R.G.; et al. Recommendations for the nomenclature of cognitive change associated with anaesthesia and surgery-2018. Br. J. Anaesth. 2018, 121, 1005–1012.
- Duning, T.; Ilting-Reuke, K.; Beckhuis, M.; Oswald, D. Postoperative delirium—Treatment and prevention. Curr. Opin. Anaesthesiol. 2021, 34, 27–32.
- Fu, D.; Tan, X.; Zhang, M.; Chen, L.; Yang, J. Association between frailty and postoperative delirium: A meta-analysis of cohort study. Aging Clin. Exp. Res. 2022, 34, 25–37.
- Needham, M.J.; Webb, C.E.; Bryden, D.C. Postoperative cognitive dysfunction and dementia: What we need to know and do. Br. J. Anaesth. 2017, 119, i115–i125.
- Shoair, O.A.; Grasso Ii, M.P.; Lahaye, L.A.; Daniel, R.; Biddle, C.J.; Slattum, P.W. Incidence and risk factors for postoperative cognitive dysfunction in older adults undergoing major noncardiac surgery: A prospective study. J. Anaesthesiol. Clin. Pharmacol. 2015, 31, 30–36.
- Borozdina, A.; Qeva, E.; Cinicola, M.; Bilotta, F. Perioperative cognitive evaluation. Curr. Opin. Anaesthesiol. 2018, 31, 756–761.
- Hermanides, J.; Qeva, E.; Preckel, B.; Bilotta, F. Perioperative hyperglycemia and neurocognitive outcome after surgery: A systematic review. Minerva Anestesiol. 2018, 84, 1178–1188.
- Badenes, R.; Qeva, E.; Giordano, G.; Romero-García, N.; Bilotta, F. Intranasal Insulin Administration to Prevent Delayed Neurocognitive Recovery and Postoperative Neurocognitive Disorder: A Narrative Review. Int. J. Environ. Res. Public Health 2021, 18, 2681.
- Neirijnck, Y.; Papaioannou, M.D.; Nef, S. The Insulin/IGF System in Mammalian Sexual Development and Reproduction. Int. J. Mol. Sci. 2019, 20, 4440.
- Oghbaei, H.; Fattahi, A.; Hamidian, G.; Sadigh-Eteghad, S.; Ziaee, M.; Mahmoudi, J. A closer look at the role of insulin for the regulation of male reproductive function. Gen. Comp. Endocrinol. 2021, 300, 113643.

