Data on the effectiveness of INI in the treatment of AD and other cognitive dysfunctions open up prospects for its use in the treatment of other pathologies, which are also, at least in part, due to functional changes in insulin signaling in the brain. These pathologies include ischemia (stroke) and brain injury, in which there is hyperactivation of apoptotic and inflammatory processes in neurons and glial cells. This leads to severe, extensive neurodegenerative changes, impaired synaptic plasticity, and disintegration of brain signaling systems. INI can be effective in diabetic pathology not only for preventing and correcting cognitive disorders caused by diabetes mellitus (DM) but also for normalizing feeding behavior, energy homeostasis, and functions of the endocrine and other systems, the regulation of which directly or indirectly depends on the brain insulin system. Significant expectations are now associated with the use of INI for the correction and prevention of CNS dysfunction caused by anesthesia, the so-called postoperative delirium, which develops in the postoperative period and significantly worsens the prognosis of surgical interventions.
2. Insulin and Insulin Signaling System in the Brain and Intranasal Delivery of Insulin to the Brain
2.1. Sources of Insulin in the Brain
The origin of insulin in the brain is associated mainly with the intake of hormone produced by β-cells from the bloodstream due to its transport through the blood–brain barrier (BBB). However, a high level of insulin in the brain during the neonatal period of development, when the insulin-producing function of the pancreas is either absent or still insufficient for the synthesis of the required amounts of the hormone, indicates the possibility of insulin synthesis in the brain de novo, at least at the early stages of ontogeny. This is evidenced by the presence of mRNA for proinsulin in a number of brain regions in rat embryos and in newborn rats and rabbits [
50], as well as in the culture of neurons obtained from various regions of the rabbit embryonic brain [
51]. In this regard, it is important to note that neurons, such as pancreatic β-cells, are electrically excitable and respond by depolarization and exocytosis to the action of hormones and to an increase in glucose levels, which is very important for the synthesis and secretion of insulin by them, since these processes require depolarization of ATP-sensitive potassium channels functionally active in both cell types [
52,
53].
The entry of insulin circulating in the blood into the brain through the BBB is carried out using three main mechanisms. The first includes the transport of insulin to brain structures using receptor-mediated endocytosis (transcytosis), which is based on the binding of the hormone to insulin receptors (INSRs) located on the surface of endothelial cells, the subsequent internalization of insulin-receptor complexes included in vesicles into cytoplasm, and their transfer to the opposite side of the endothelial cell, followed by exocytosis on its abluminal side and the release of insulin towards the endings of neurons or glial cells [
54,
55]. The decisive role of INSR in this process is evidenced by the fact that in mice with a knockout of the
Insr gene in endothelial cells, the permeability of the BBB for insulin deteriorates sharply, and as a result, insulin signaling in the hypothalamus, hippocampus, and prefrontal cortex is impaired [
56].
There is competition between insulin and insulin-like growth factor-1 (IGF-1), another member of the insulin family, for receptors that mediate the transport of insulin across the BBB, since IGF-1 at higher than physiological concentrations is able to suppress insulin transcytosis [
57]. Insulin transcytosis is regulated by hormones, lipids, and vasodilation factors. It significantly depends on the physiological parameters, weakens with obesity and MS, and increases with inflammatory diseases and insulin-deficient forms of DM, although the mechanisms in each case differ and are not always investigated [
55,
58,
59,
60,
61,
62,
63,
64,
65]. Astrocytes and pericytes, which form close contacts with vascular endothelial cells, play an important role in modulating transendothelial insulin transport across the BBB [
66]. The loss of pericytes or a violation of their structure leads to an increase in the permeability of the BBB and an impaired selectivity of transport of insulin and other hormones through the BBB [
67,
68,
69].
Other mechanisms include passive insulin transport without INSR involvement through the fenestrated capillaries and ependymal cells of the median eminence, located below the mediobasal hypothalamus, ventral to the third ventricle and adjacent to the arcuate nucleus of the hypothalamus [
70]. The transport of insulin across the median eminence is regulated by tanycytes, highly specialized ependymal cells that line the ventrolateral wall and the floor of the third ventricle and are involved in the exchange of substances between the blood and cerebrospinal fluid [
71,
72]. The presence of tight junctions between adjacent tanycytes provides a physical barrier that controls the transfer of insulin from portal capillaries and cerebrospinal fluid to the hypothalamus [
73,
74]. Remodeling of the hemato-hypothalamic barrier, for example, due to changes in blood glucose levels, affects the transfer of insulin and other circulating hormones through it to the arcuate nuclei of the hypothalamus [
73,
75]. In this regard, it should be noted that insulin enters the arcuate nuclei of the hypothalamus mainly through its passive transport with the participation of tanycytes, and not through receptor-mediated transcytosis [
72].
2.2. Insulin Signaling System
A key component of the insulin signaling system in brain neurons and glial cells are INSRs, which are found in the hypothalamus, hippocampus, cerebellum, thalamus, cerebral cortex, olfactory bulb, dentate gyrus, motor cortex, and other regions [
76,
77,
78,
79,
80,
81,
82,
83,
84,
85]. In mammals, two INSR isoforms are present, the full-length (INSR-B) and the truncated (INSR-A). INSR-A is formed as a result of alternative splicing of the 11th exon by removing a 12 amino acid segment from the encoded sequence. These INSR isoforms are co-expressed in many tissues, but the ratio of INSR-A/INSR-B in the brain is much higher than at the periphery, which is why INSR-A is often referred to as the neuronal isoform of the receptor [
86,
87,
88]. INSR-A and INSR-B are similar in structural organization and pharmacological characteristics, although they have a number of functional differences, including those due to differences in glycosylation [
89,
90,
91]. Insulin is also able, albeit with low affinity, to bind to a hybrid receptor that includes the monomers of both INSR and IGF-1 receptor (IGF1R). The IGF1R is structurally and functionally similar to INSR, since the genes encoding these receptors,
Igf1r and
Insr, originate from a common ancestral gene and are part of an ancient, highly conserved signaling system in vertebrates and invertebrates involved in the regulation of cell metabolism, growth, and differentiation [
92,
93].
Both INSR isoforms are α
2β
2-heterotetramers in which extracellular α-subunits and transmembrane β-subunits are linked by disulfide bonds [
94,
95]. The α-subunit and the N-terminal segment of the β-subunit form the INSR ectodomain responsible for high-affinity insulin binding. The large cytoplasmic domain of the β-subunit contains a highly conserved tyrosine kinase domain that is stimulated after insulin binds to the ligand-binding site located in the α-subunit [
94]. Binding of the α-subunit to insulin results in conformational rearrangements that affect the tyrosine kinase domain of the β-subunit and induce receptor autophosphorylation at the tyrosine residues, Tyr
1158, Tyr
1162 and Tyr
1163, the main targets of the receptor tyrosine kinase. After autophosphorylation, the β-subunit interacts with regulatory and adapter proteins that contain phosphotyrosine-binding sites that can specifically interact with segments including phosphorylated Tyr
1158, Tyr
1162 and Tyr
1163 (
Figure 1). Depending on the pattern of this interaction, various signaling pathways are activated, resulting in different responses of the target cell to insulin [
96].
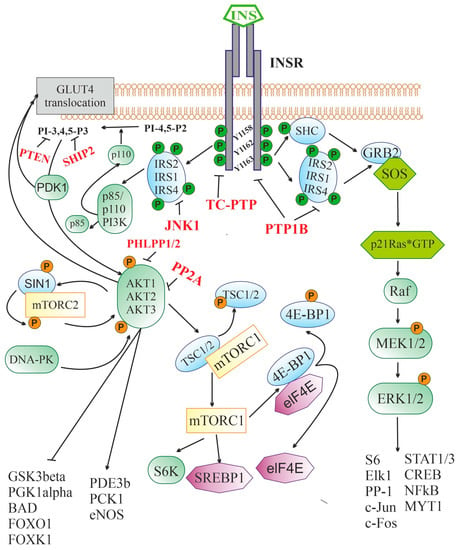
Figure 1. Insulin-activated signaling pathways. Abbreviations: INSR—insulin receptor, containing three sites for tyrosine phosphorylation (Y1158, Y1162, and Y1163); IRS1, IRS2, and IRS4—insulin receptor substrates-1, -2 and -4, respectively; p85 and p110 PI3K—p85-regulatory and p110-catalytic subunits of phosphatidylinositol-3-kinase; PI-4,5-P2—phosphatidylinositol-4,5-diphosphate; PI-3,4,5-P3—phosphatidylinositol-3,4,5-triphosphate; PDK1—phosphoinositol-dependent protein kinase-1; AKT1, AKT2, and AKT3—serine/threonine-specific protein kinase B (AKT kinase) of the types 1, 2 and 3, respectively; DNA-PK—DNA-dependent protein kinase; mTORC2—mammalian target of rapamycin complex 2 that includes the protein kinase mTOR, the regulatory protein RICTOR (Companion of mammalian Target Of Rapamycin), mSIN1 protein (mammalian Stress-activated protein kinase Interacting Protein 1) and some other components; GLUT4—type 4 insulin-dependent glucose transporter; GSK3beta—glycogen synthase kinase-3β; PGK1alpha—peroxisome proliferator-activated receptor-gamma coactivator 1α; BAD—BCL2 antagonist in cell death; FOXO1 and FOXK1—transcription factors of FOX (forkhead box) family; PDE3B—subtype 3B cAMP-dependent phosphodiesterase; PCK1—phosphoenolpyruvate carboxykinase 1; eNOS—endothelial isoform of NO synthase; mTORC1—mammalian target of rapamycin complex 1; TSC1/2—tuberous sclerosis proteins 1 (hamartin) and 2 (tuberin); S6K—p70 ribosomal S6 kinase; SREBP1—Sterol regulatory element-binding transcription factor 1; eIF-4E—eukaryotic translation initiation factor 4E; 4E-BP1—eukaryotic translation initiation factor 4E-binding protein 1; SHC—adapter SH2/α-collagen-like protein; GRB2—adapter protein-2 associated with growth factor receptors; SOS—metabolic protein (Son of Sevenless), induced GDP/GTP exchange; p21Ras*GTP—small GTP-binding protein of the Ras family in GTP-bound form; Raf—serine/threonine-specific protein kinase; MEK1/2—mitogen-activated protein kinase kinases-1 and -2; ERK1/2—mitogen-activated protein kinases-1 and -2; Elk-1—transcription factor containing the ETS (E26 transformation-specific) domain; STAT1/3—types 1 and 3 signal transducers and transcription activators; CREB—cAMP-dependent transcription factor; S6—ribosomal protein S6; NF-κB—nuclear factor κ-light-chain-enhancer of activated B cells; MYT1—myelin transcription factor 1; c-Fos and c-Jun—the transcription factors of the Fos and Jun families, respectively; PP-1—protein phosphatase 1; PTEN—phosphatase (Phosphatase and TENsin homolog) hydrolyzing PI-3,4,5-P3 to PI-4,5-P2; SHIP-2—SH2 domain-containing inositol-5’-phosphatase 2 hydrolyzing PI-3,4,5-P3 to PI-3,4-P2; JNK1—c-Jun N-terminal kinase-1; PP2A—protein phosphatase 2A subtype; PHLPP1/2—plekstrin-homologous (PH)-domain leucine-rich-repeat-containing protein phosphatases; PTP1B—protein phosphotyrosine phosphatase 1B; TC-PTP—T-cell protein phosphotyrosine phosphatase.
After binding to the extracellular domain of INSR, insulin stimulates the intracellular receptor tyrosine kinase, which leads to the phosphorylation of three tyrosine residues (Y1158, Y1162, and Y1163), and further to the tyrosine phosphorylation of the IRS proteins and the adapter SHC protein. Through IRS proteins, the 3-phosphoinositide pathway is stimulated, including the enzyme PI3K catalyzing the synthesis of PI-3,4,5-P3 from PI-4,5-P2 and the effector serine/threonine protein kinase AKT. The targets of AKT-kinase are numerous enzymes and transcription factors that control cell growth, apoptosis, differentiation and metabolism, and AKT-kinase inhibits some of them (GSK3β, PGK1α, BAD, transcriptional factors of the FOX family), while others stimulate (PDE3b, PCK1, eNOS). AKT activates the protein kinase complex mTORC1, which leads to the stimulation of p70S6K kinase and a number of transcription factors involved in the regulation of protein and lipid synthesis. Along with this, AKT and PDK1 are also involved in the process of translocation of the insulin-dependent glucose transporter GLUT4 into the cell membrane, followed by GLUT4-mediated glucose uptake. Through tyrosine-phosphorylated IRS or SHC, insulin triggers a cascade of mitogen-activated protein kinases. The SHC protein in phosphorylated form is able to form a complex with GRB2 and SOS, which makes it possible to induce the exchange of guanine nucleotides in small G protein, p21Ras, converting it into an active GTP-bound form. Downstream components of the cascade of mitogen-activated protein kinases, ERK1/2 kinases, phosphorylate and thereby stimulate the activity of many transcription factors and enzymes, regulating the transcriptional activity of the genome. The functional activity of the insulin signaling cascade is under the control of a large number of negative regulators, the targets of which are INSR (PTP1B and TC-PTP), IRS proteins (PTP1B and JNK1), 3-phosphoinositides (PTEN, SHIP2), and AKT-kinase (PP2A and PHLPP1/2).
Insulin receptor substrates (IRS) proteins, which contain a large number of phosphotyrosine-binding sites, play a leading role in the transduction of the insulin signal from the receptor to intracellular effectors. The interaction of INSR with them activates several signaling pathways at once [
96]. Despite a significant number of IRS protein isoforms, the IRS-1 and IRS-2 isoforms, which are ubiquitously expressed, are of the greatest importance for insulin signaling. At the periphery, IRS-1 is more involved in insulin signal transduction, while, in the CNS, the situation is different, and IRS-2 is most relevant for insulin signal transduction [
97]. There is evidence that in the brain, the IRS4 proteins are involved in the transduction of signals generated by insulin and IGF-1. IRS4 can functionally replace IRS-1 and IRS-2, and in hypothalamic neurons, together with IRS-2, they are involved in the regulation of glucose homeostasis, feeding behavior, and energy expenditure [
98,
99]. In the N-terminal part of IRS proteins, the plekstrin-homology and phosphotyrosine-binding domains are localized, which ensure their interaction with INSR and association with the plasma membrane. The C-terminal part of IRS proteins contains a domain involved in specific interaction with various types of src-homology-2 (SH2)-domain-containing proteins responsible for the cell’s response to insulin [
100]. Among the SH2-domain-containing proteins that are targets of insulin and IGF-1, the most important are phosphatidylinositol-3-kinase (PI3K), adapter protein GRB2, protein phosphotyrosine phosphatase SHP2, non-receptor tyrosine kinase Fyn, and suppressors of cytokine signaling.
After interacting with activated INSR, IRS proteins acquire the ability to bind to the phosphotyrosine-binding domain of the p85 regulatory subunit of PI3K and release the catalytic subunit of the enzyme, p110, which converts phosphatidylinositol-4,5-diphosphate to phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3), the most important second messenger [
101] (
Figure 1). PI-3,4,5-P3 induces the translocation of 3-phosphatidylinositol-dependent protein kinases of types 1 and 2 (PDK1/2), SIN1 protein, a component of the mammalian target of rapamycin complex 2 (mTORC2), and various isoforms of serine/threonine protein kinase AKT (AKT1, AKT2, or AKT3) to the plasma membrane to form an oligomeric complex, resulting in the activation of AKT [
96]. In the CNS, the expression of AKT kinase isoforms is specific to both certain cell types and brain regions. AKT1 and AKT3 are mainly expressed in neurons, and, to the greatest extent, in the amygdala, hippocampus, and cerebral cortex, while AKT2 is predominantly expressed in astrocytes [
102]. Membrane-bound PDK1 phosphorylates AKT at Thr
308, while mTORC2 and DNA-dependent protein kinase (DNA-PK) at Ser
473, and phosphorylation at two sites at once leads to complete activation of the enzyme [
103]. The targets of AKT are phosphodiesterase 3B, phosphoenolpyruvate carboxykinase 1, and endothelial NO synthase, which are stimulated by AKT-induced phosphorylation, as well as peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α), glycogen synthase kinase-3β (GSK3β), and proapoptotic protein BAD, whose activity is reduced after AKT phosphorylation. AKT-induced phosphorylation of transcription factors of the forkhead box family (FOXO1, FOXK1) inhibits their activity and suppresses FOX-dependent gene transcription [
96].
AKT kinase plays an important role in the control of cellular functions by regulating the expression of the genes responsible for cell growth, survival, differentiation, and metabolism. Along with this, it is involved in the translocation of the glucose transporter GLUT4, which provides insulin-dependent glucose uptake by cells into the plasma membrane [
104]. AKT-induced phosphorylation of GSK3 at Ser
21 (GSK3α) or Ser
9 (GSK3β) results in GSK3 inactivation and blocks GSK3-mediated regulation of glycogen synthase activity, a key enzyme in glycogen synthesis that controls glucose homeostasis [
105]. Along with the glycogen synthesis, GSK3 regulates the activity of a large number of transcription factors and their coactivators and corepressors, including NF-κB, Snail, Notch, BAD, and transcription factors of the FOX family. As a result, AKT-mediated inhibition of GSK3 makes a significant contribution to the effects of insulin on gene expression, apoptosis, autophagy, and cell differentiation [
105,
106,
107,
108]. In the CNS, GSK3β controls neuronal progenitor cell proliferation, neuronal polarity, and neuroplasticity [
109,
110]. GSK3β phosphorylates the tau protein, hyperphosphorylated forms of which are associated with neurodegenerative diseases, including AD. As a consequence, the weakening of insulin signaling in the brain, for example, in the conditions of neuron-specific knockout of INSR and IRS-2, promotes hyperphosphorylation of the tau protein and provokes neurodegeneration and cognitive deficit [
111]. In addition, as noted above, transcription factors of the FOX family, which control various functions in the CNS, including energy homeostasis, feeding behavior, and locomotor activity, are the most important target of GSK3β [
112,
113]. The dysregulation of FOX-family factor activity by insulin, mediated through the 3-phosphoinositide pathway and GSK3β, leads to neurodegenerative diseases and metabolic disorders [
114,
115].
Another target of insulin is the mitogen-activated protein kinase (MAPK) cascade, which is stimulated by insulin through a signaling pathway including activated INSR, IRS proteins, GRB2 adapter protein, guanine nucleotide exchange factor SOS (Son of Sevenless), and p21ras, a small G protein (
Figure 1). The GRB2/SOS complex stimulates the exchange of guanine nucleotides in p21ras, converting it into an active GTP-bound form, which triggers the MAPK cascade: Ras-protein→Raf-kinase→MEK1/2→ERK1/2. ERK1/2 activation and results in the increased activity of transcription factors and regulatory proteins, such as ribosomal protein S6, NF-κB, protein phosphatase-1 (PP-1), MYT1, Elk-1, cAMP-regulated transcription factor CREB, proto-oncogenes c-Fos and c-Jun, and transcription factors STAT family, which control a wide range of cellular processes [
96,
116]. Since the MAPK cascade is also regulated by other hormonal stimuli, at the level of various MAPKs, there is a crosstalk between insulin and other signaling systems, both in the brain and at the periphery.
AKT also phosphorylates TSC1/2 adapter proteins, and tuberous sclerosis proteins 1 (hamartin) and 2 (tuberin), which are negative regulators of mammalian target of rapamycin complex 1 (mTORC1). The result of this phosphorylation is the abolition of the blocking effect of TSC1/2 on the kinase activity of mTORC1 (
Figure 1). Activated via AKT, mTORC1 controls the activity of factors that induce autophagy, as well as ribosomal p70-S6 kinase and transcription initiation factor eIF4E, which makes a significant contribution to the regulation of ribosome biogenesis, the processes of transcription, translation, and degradation of proteins [
96,
117]. mTORC1-dependent protein synthesis is extremely important for providing neuronal plasticity, as well as for the formation and maintenance of integrative connections between various brain structures [
118]. In addition, mTORC1 activation enhances lipid synthesis through the activation of SREBP1 (Sterol regulatory element-binding transcription factor 1), which provides insulin control of lipid metabolism [
119].
INSR activation, along with stimulation of intracellular signaling cascades, leads to the activation of negative feedback mechanisms that prevent hyperactivation of the insulin system and return it to its initial inactive state. Along with the stimulation of endocytosis of insulin-receptor complexes with their subsequent recycling or degradation in proteasomes, inactivation of the upstream components of the insulin signaling system is carried out by changing the phosphorylation of INSR and IRS proteins. A significant role in this is assigned to the dephosphorylation of tyrosine-phosphorylated forms of INSR and IRS proteins by tyrosine phosphatases, such as protein phosphotyrosine phosphatase 1B (PTP1B) and T-cell protein phosphotyrosine phosphatase (TC-PTP) [
120,
121,
122]. Along with this, the activity of IRS proteins can be inhibited by their phosphorylation at serine and threonine residues with c-Jun N-terminal kinase-1 (JNK1) [
123]. Thus, an increase in the expression and functional activity of phosphatases PTP1B and TC-PTP and JNK1 kinase, which reduces the efficiency of insulin signaling, is one of the causes of peripheral and central IR, including in obesity, MS, and T2DM [
121,
124]. In hyperphagia and obesity, hyperactivation of both tyrosine phosphatases is observed in the hypothalamus, which leads to a decrease in the sensitivity of hypothalamic neurons to insulin, and in prolonged consumption of saturated fatty acids, JNK1 activity increases in the hypothalamus and other regions of the brain, which reduces the activity of IRS proteins and leads to central IR [
124,
125]. A decrease in JNK1 activity prevents the negative effects of a long-term high-fat diet on insulin sensitivity in the brain, as well as on insulin-mediated metabolic regulation and cognitive functions [
126].
Along with the suppression of the activity of INSR and IRS proteins, the negative regulation of insulin signaling can be carried out for its downstream effector components, as in the case of the 3-phosphoinositide pathway. Several phosphatases can act as negative regulators of this pathway, such as protein phosphatase PP2A, phosphatase PTEN (Phosphatase and TENsin homolog), and protein phosphatases PHLPP-1 and PHLPP-2 (PH domain and Leucine-rich repeat Protein Phosphatases 1/2) [
104]. Phosphatase PTEN prevents the synthesis of 3-phosphoinositides by hydrolyzing PI-3,4,5-P3, while phosphatases PP2A and PHLPP1/2 dephosphorylate AKT kinase, inhibiting its activity. Another negative regulator of the 3-phosphoinositide pathway is type 2 SH2-containing inositol-5’-phosphatase (SHIP2), whose expression has been shown in various regions of the brain [
127]. It has been shown that the level of PTEN in the substantia nigra of patients with Parkinson’s disease is significantly increased, which is associated with enhanced neurodegeneration [
128], and an increase in the content of PTEN increases brain damage after ischemia/reperfusion [
129]. Hyperactivation of phosphatase PP2A in the brain, causing central IR and an imbalance in the regulatory effects of insulin and other neurohormones, is closely associated with neurodegenerative diseases, including AD and Parkinson’s disease [
130].
It should be noted that all the main blocks of the brain insulin signaling system are involved in the implementation of the neuroprotective, neurotrophic, and neuromodulatory effects of INI and also mediate its effects on other hormonal and neurotransmitter systems that are integrated and coordinated with the brain insulin system.
2.3. The Intranasal Route of Insulin Delivery
Over the past decades, the intranasal route of administration has become relevant for the delivery of drugs to the CNS, bypassing the BBB. More than 30 years ago, William H. Frey II was the first to propose the use of intranasal administration of various biologically active substances, including polypeptide hormones and growth factors, for the treatment of Alzheimer’s disease and other diseases of the nervous system, and insulin was of the greatest interest among the studied hormones [
131,
132,
133]. To date, there are already more than 50 clinical trials of INI in both healthy subjects and patients with CNS diseases and diabetes (
https://clinicaltrials.gov/, accessed on 27 December 2022).
The main advantage of intranasal administration of drugs is the targeted delivery to the CNS of substances that are unable to penetrate the BBB due to their size, charge, or other physical, chemical and biological properties. This approach provides a rapid achievement of a therapeutic effect, as well as non-invasiveness and ease of use compared to oral or injection methods of drug delivery. An important feature of intranasal administration is the absence of passage through the hepatic barrier, which allows the use of lower doses of drugs that reduce side effects [
16,
20,
21].
Significant progress in the study of the INI mechanisms involved in bypassing the BBB, its pharmacokinetics and pharmacodynamics, as well as the targets of INI in the brain, has been achieved through the use of animal models [
16,
17,
18,
19,
134]. After nebulization in the nasal cavity, insulin enters the nasal mucosa, where it is transported to the brain by an intracellular route using receptor-mediated endocytosis via axons of olfactory and trigeminal nerves, which innervate the nasal cavities, as well as via extracellular pathway by interstitial fluid, but with different speeds. Retrograde axonal transport is rather slower [
135] than an extracellular one, so it takes hours to reach the inner brain structures, and more likely that INI is primarily transported along extra-neuronal routes [
16,
136]. The extra-neuronal pathway rapidly allows insulin and its homolog IGF-1 to spread along the olfactory and trigeminal nerves and distribute within brain in the rostral to caudal direction [
18,
19,
137]. Once in the CNS, insulin binds to brain INSRs and causes their activation, which is demonstrated by an increase in the level of tyrosine receptor phosphorylation [
19]. This leads to activation of intracellular signaling pathways, including the PI-3-K/Akt signaling pathway, as mentioned above [
17,
18]. After 15–30 min after intranasal administration, insulin accumulation was shown in the olfactory bulbs, striatum, substantia nigra, brainstem, cerebellum, and, to a lesser extent, in the hippocampus and cerebral cortex [
17,
18]. According to data from other authors, large amounts of intranasally administered insulin were found in the cortex, cerebellum, hippocampus, and hypothalamus [
19]. Thereafter, the amount of insulin in the brain gradually decreases but remains elevated even 6 h after its administration [
18]. An increase in insulin levels after intranasal administration was also found in the cerebrospinal fluid and in a small amount in the serum [
134,
138,
139]. With long-term (9 days) administration to mice, INI increased the level of glucose, adenosine triphosphate, and phosphocreatine in the brain of animals, which indicates its positive effect on energy metabolism in the brain [
134]. In this regard, it should be noted that in humans, a positive effect of INI on improving glucose homeostasis and energy metabolism in the brain has been demonstrated, which is important for correcting hypometabolic conditions in neurodegenerative and other diseases [
140].
In human studies, INI bypassed the BBB and detected in the cerebrospinal fluid at 10 min with peak at 30 min and remained significantly elevated at 80 min [
24]. The serum insulin levels peaked in 10–20 min after its intranasal administration, then remained slightly elevated for approximately 1 h and returned to mean values 1.5–2 h after nebulization with an insignificant risk of hypoglycemia mainly in a fasting state [
25,
141]. The fast and unhindered entry of INI into the CNS is a great advantage of the intranasal route of hormone administration compared to other routes of its administration, which is important in pathologies that require emergency treatment, such as stroke and brain injury, as well as during pre- and postoperative procedures.
The data obtained from the use of INI in humans does not raise any significant concerns in its short-term and long-term use. On the contrary, most studies point to its positive effect on cognitive functions, good tolerability, and the absence of serious side effects that could become an obstacle to INI therapy. However, it should be noted that the chronic, long-term use of INI in a small cohort of patients still revealed some undesirable effects, which are mainly due to the procedure of spraying INI in the nasal cavity (rhinitis, mild epistaxis, sneezing, headaches, and predisposition to respiratory tract infections) [
26,
27,
28,
33,
46]. It is important that these undesirable effects can be eliminated by optimizing both the procedure for intranasal nebulization of the hormone and the composition of the mixture for nebulization, since they are not caused by the toxic effect of INI on the nasal cavity and brain structures.
3. Intranasal Insulin and Brain Ischemia
Cerebral ischemia occurs as a result of various critical events and diseases, including surgery, resuscitation, traumatic brain injury, asphyxia, hemorrhages, embolisms, myocardial infarction, acute blood loss, and hypotension [
142,
143,
144]. However, the main cause of damage to brain neurons is a deficiency of oxygen and nutrients in the CNS. The consequences of cerebral ischemia are determined by the level of damage and death of neurons and glial cells and depend on the degree of deterioration of blood flow in the brain tissue, the duration of the ischemic episode, and the area of the ischemic lesion. The metabolic and hormonal status of the brain and organism before cerebral ischemia is also of great importance for the severity of CNS damage caused by it [
143]. Developing glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, inhibition of protein synthesis, and activation of proteolytic enzymes lead to the triggering of endoplasmic reticulum stress, cell death, and neuroinflammation. Prolonged brain ischemia leads to disintegration and irreversible changes in the brain signaling cascades responsible for the survival and functional activity of neurons, resulting in their death through apoptosis, necrosis, or autophagy [
145]. Not only neurons but also glial cells are damaged during ischemia, which negatively affects the neuroprotective potential of glia. At the brain level, cerebral ischemia is accompanied by cerebral edema, damage to the integrity and permeability of the BBB, hyperactivation of microglia, and increased pro-inflammatory cascades [
146,
147,
148,
149]. During ischemia, compensatory mechanisms are activated that maintain the viability of neurons and reduce the intensity of pro-inflammatory and apoptotic processes in the CNS. These mechanisms include signaling cascades triggered by a large number of neurohormones, growth factors and cytokines with neuroprotective properties, and the insulin and IGF-1 signaling cascades are of key importance among them [
145]. As a result, the use of hormonal regulators with neuroprotective properties, including insulin, is able to compensate for ischemic brain damage, which makes them promising drugs for the treatment of ischemia.
Despite many years of research in the field of treatment of ischemic stroke, there are currently no anti-ischemic neuroprotective drugs that are used clinically [
150,
151]. Since the pathophysiology of stroke is multi-faceted, it is promising to use neuroprotectors with a pleiotropic effect, which makes it possible to prevent several pathological processes in the brain at once. This predetermines the main direction of the search for pharmacological agents to treat postischemic dysfunctions of the nervous and other systems.
Hyperglycemia leads to more severe consequences [
152,
153,
154], making glycemic control one of the most important steps in the treatment and rehabilitation of patients with cerebral ischemia [
155]. In this regard, it should be noted that both hyperglycemia and hypoglycemia, including those induced by insulin therapy, are associated with a poor prognosis of ischemia [
156,
157]. In this regard, it is not surprising that patients with DM or pre-diabetes have an increased risk of ischemic and hemorrhagic stroke [
158,
159,
160,
161]. Along with this, about half of the patients who had an ischemic stroke later had impaired glucose homeostasis and IR [
162,
163]. T2DM increases the risk of recurrent stroke after the first episode of ischemia or stroke, so secondary prevention of stroke in patients with metabolic disorders is also an important problem [
150,
151]. There is evidence that weight loss, restoration of tissue sensitivity to insulin, and normalization of glucose homeostasis are very effective in stroke prevention [
150,
151,
158,
159,
160,
161].
All of the above indicates that injectable forms of insulin, which normalizes glucose levels in hyperglycemia, can be indicated for the correction of hyperglycemic conditions in diabetic patients with stroke, as well as in non-diabetic patients with impaired glucose homeostasis that developed as a result of a stroke. However, in the recent years, the focus of the use of insulin has shifted from its metabolic effects to neuroprotective effect and the ability of insulin to influence the functional activity of brain structures that have undergone ischemic damage. In connection with the foregoing, let us consider the currently available data on the effect of insulin and its functional homolog, IGF-1, on postischemic damage in animals with experimental ischemic and hemorrhagic stroke and hypoxia-ischemia.
The neuroprotective properties of insulin in ischemic injury have been studied for more than 30 years [
164,
165,
166,
167]. It has been shown that insulin with various delivery methods (subcutaneous, intraperitoneal, intravenous, and intracerebroventricular) positively affects the survival and cognitive functions of animals that have undergone cerebral ischemia [
165,
168,
169,
170,
171,
172,
173,
174,
175,
176]. The antiapoptotic signaling pathway, including IR/IRS/PI3K/AKT, plays a leading role in the implementation of the neuroprotective action of insulin. By activating AKT, insulin suppresses the release of cytochrome
C from mitochondria, thereby preventing the translocation of the proapoptotic protein BAX to them [
177,
178], and also stimulates synthesis of neuroprotective proteins, including the antioxidant enzymes [
179,
180], which increases the survival of neurons and prevents cognitive dysfunctions in animals with cerebral ischemia [
178]. The contribution of insulin to the improvement of cognitive functions during oxidative stress or excitotoxicity may also be due to its neuromodulatory effect, by changing the surface density and activity of the glutamate and γ-aminobutyric acid ionotropic receptors in neurons, as well as by regulating the membrane transport of glutamate and γ-aminobutyric acid, which allows optimizing their ratio in the extracellular space [
180].
The vasoactive effect of insulin is widely known, which, along with its neuroprotective effect, plays an important role in the implementation of the protective functions of insulin in cerebral ischemia. AKT-dependent activation of endothelial NO-synthase under the action of insulin on the endothelium of cerebral vessels leads to changes in the vascular lumen and improves the cerebral blood flow (CBF) [
181,
182]. At the same time, depending on the dose of the hormone and the functional state of the CNS, insulin is able to both increase and decrease the lumen of the vessels, exerting a modulating effect on CBF [
183,
184]. In healthy subjects, an increase in insulin levels in the brain leads to increased CBF and improved blood supply to brain tissue [
185].
Despite a significant number of studies on the effects of peripheral and intracerebroventricular insulin administration to correct the consequences of cerebral ischemia, there are relatively few data on the use of INI, despite the promise of this method for correcting ischemic damage [
186,
187,
188,
189,
190,
191,
192]. It should be noted that intranasally administered IGF-1 has been used for the correction of damage caused by cerebral ischemia–reperfusion for 20 years [
186,
193,
194,
195,
196,
197,
198,
199]. The targets of action of insulin and IGF-1 in the CNS largely coincide, which is due to both the similar architecture of their signaling cascades and the ability of insulin and IGF-1 to activate the same receptors, including hybrid di/oligomeric forms of INSR/IGF1R [
95]. In addition, both hormones have a similar pattern of regulatory effects on neurons and glial cells, exerting antiapoptotic and anti-inflammatory effects, as demonstrated for insulin in the use of INI for the treatment of neurodegenerative diseases, including AD [
27,
200,
201]. All this allowed the group of Vera Novak to put forward a hypothesis about the prospects of using INI to correct cerebral ischemia [
186]. It is important that INI has clear advantages over intranasally administered IGF-1, because, unlike insulin, IGF-1 is characterized by a very powerful mitogenic effect, which has significant risks of cell malignancy, and the IGF-1 dosage form is expensive and has very limited clinical experience. It is also important that INI not only has a neuroprotective effect on neurons and glial cells but also improves the brain blood supply, which is especially valuable in the conditions of cerebral ischemia [
185]. The potential mechanisms of neuroprotective action of INI in cerebral ischemia are shown in
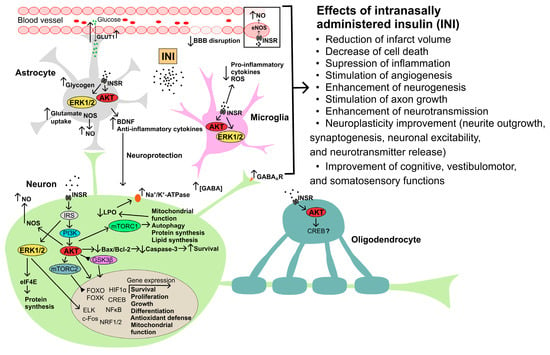
Figure 2.
Figure 2. The possible mechanisms of neuroprotective action of intranasally administered insulin in cerebral ischemia and traumatic brain injury. Abbreviations: AKT—AKT kinase; Bax—Bcl-2-associated X protein, proapoptotic factor; BBB—blood–brain barrier; Bcl-2—B-cell lymphoma 2 protein, antiapoptotic factor; BDNF—brain-derived neurotrophic factor; c-Fos—transcription factor of the FOS family; CREB—cAMP response element-binding protein; eIF4E—eukaryotic initiation factor 4E; ELK—ETS Like-1 protein Elk; ERK1/2—extracellular signal-regulated kinases-1 and -2; FOXO and FOXK—forkhead box transcription factors of the O and K subfamilies; GABA—γ-aminobutyric acid; GABAAR—ionotropic GABAA receptor; GLUT1—type 1 glucose transporter; GSK3β—glycogen synthase kinase-3β; HIF1α—hypoxia-induced factor-1α; INSR—insulin receptor; IRS—insulin receptor substrate; LPO—lipid peroxidation; mTORC1, mTORC2—mammalian target of rapamycin complexes 1 and 2, respectively; NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells; NO—nitric oxide; NOS—NO synthase; NRF1/2—nuclear respiratory factors 1 and 2; ROS—radical oxygen species.
4. Intranasal Insulin and Brain Injury
Traumatic brain injury (TBI) in most cases leads to long-term cognitive deficit, which is currently poorly or not treatable at all [
238]. Even mild TBIs in 15% of patients lead to cognitive deficits in the long term [
239]. TBI is based on a three-phase response of the brain to an external damaging effect [
240]. The first phase corresponds to the development immediately after injury of a hypermetabolic state caused by a powerful release of ions and glutamate, which is due to a sharply increased demand for energy. The second phase corresponds to a significant decrease in the absorption of glucose by the brain tissue, which leads to an energy deficit in neurons and is the main cause of brain damage and triggering neurodegenerative processes. The long-term consequences of TBI, including the subsequently developing cognitive deficit, largely depend on the duration and severity of this phase. The final phase includes a gradual return of glucose uptake by the brain to normal values [
240]. It is important that the restoration of metabolism can occur only in some regions of the brain, while other regions are characterized by a decrease in metabolic status over a long period, years after TBI, even in patients with a mild form of this injury [
241,
242]. The disintegration of functional connections between various brain structures, the decrease in neuronal plasticity in them, and the activation of neuro-inflammatory processes lead to dysfunctions of the CNS and an imbalance of metabolic processes at the periphery [
243,
244,
245].
In animals with experimental TBI, a decrease in brain sensitivity to insulin was found, which may be a consequence of glutamate excitotoxicity induced by traumatic exposure and activation of pro-inflammatory processes in the brain [
244,
246,
247]. With a single traumatic impact, a decrease in the brain sensitivity to insulin is observed within 7 days, and with repeated trauma, up to 28 days [
246]. At the same time, in obese mice, which initially had impaired insulin signaling in the brain, the consequences of TBI were expressed to a much greater extent. They were accompanied by a significant weakening of the stimulatory effect of insulin on the INSR/IRS/PI3K/AKT signaling pathway, pronounced neuroinflammation, a significant deterioration in learning ability, a sharp decrease in memory, and a severe anxiety-depressive state [
246].
A study of glucose uptake in the brain of rats showed that after a mild TBI, its changes begin to be registered as early as 3–6 h after the traumatic impact and persist to some extent for 5–10 days, and this effect was specific both for certain regions of the brain and for certain types of neurons and glial cells [
248,
249,
250]. The development of post-traumatic complications is largely due to insulin sensitivity of the brain, which decreases with obesity and a number of other disorders, as well as the effectiveness of integrative interactions between brain regions, which can significantly affect insulin sensitivity and glucose uptake by neurons and glial cells [
244,
250]. Since after a single TBI, there is a transient weakening of the sensitivity of certain regions of the brain to insulin, it seems quite logical that repeated TBIs aggravate the central IR, especially when they occur in the time window of a decrease in insulin sensitivity caused by previous TBI. In 2022, a model of lateral fluid-percussion injury in rats showed that 4 months after traumatic exposure, glucose uptake was reduced in the hippocampus, ipsilateral striatum, and frontoparietal cortex [
251]. Decreased glucose uptake in the stratum was positively correlated with deterioration in spatial memory [
251].
All of the above points to the potential ability of INI to improve glucose homeostasis in the brain and compensate, at least in part, for the hypometabolic state that develops after TBI [
244]. This is supported by the data on INI effect on adult male Sprague Dawley rats with moderate controlled cortical injury. Treatment of animals with INI (Humulin R-100, 6 IU/rat) was started 4 h after traumatic exposure, and then INI treatment at the same dose was continued for two weeks [
252]. As a result, glucose utilization was normalized in the hippocampus of rats treated with INI, which was accompanied by a decrease in the hippocampal lesion area, a decrease in neuroinflammation mediated by microglia, and an improvement in memory and spatial orientation [
252].
The prospects for the use of INI in TBI are also indirectly evidenced by the fact that moderate and severe TBI is significantly associated with AD, which is one of the targets of INI [
253]. The severity of brain damage is positively correlated with the incidence of AD. It should be noted that a close relationship is also observed between AD and Parkinson’s disease, which have many common molecular causes, including metabolic changes in the brain that provoke neurodegeneration, and both are therapeutic targets for INI [
254]. It should also be noted that the pathogenetic mechanisms of stroke and TBI are quite similar, and therefore the previously described neuroprotective effects of INI for cerebral ischemia can also be manifested in TBI [
255]. The possible mechanisms and targets of neuroprotective action of INI in TBI are summarized in
Figure 2. Thus, the general pathogenetic factors of CNS diseases may a priori suggest the potential effectiveness of INI for the treatment of TBI and the prevention of its negative consequences.
5. Intranasal Insulin and Diabetes Mellitus
DM is characterized by significant changes in the functional activity of the insulin signaling system, and this is due to both absolute or relative insulin deficiency in type 1 DM (T1DM) and the development of IR in T2DM. In both cases, the result is a weakening of insulin signaling pathways both in the brain and at the periphery. To restore them, pharmacological approaches can be used to compensate for insulin deficiency (insulin replacement therapy) or to reduce IR (metformin and other drugs that increase insulin sensitivity). Since insulin in the brain is involved in the regulation of the functions of neurons and glial cells, controls the integrative relationships between different brain regions, and mediates the central regulation of physiological and metabolic processes at the periphery, an impaired brain insulin signaling in DM leads to a wide range of disorders. As a result, the use of INI, which activates the insulin system in the brain, may be useful for preventing cognitive deficits in patients with DM, as well as for restoring the central insulin-mediated regulation of peripheral metabolism and physiological functions.
Among the factors that lead to the development of neurodegenerative processes and cognitive deficit in T2DM and MS, the most important are cerebrovascular dysfunctions [
256,
257], reduced glucose metabolism in brain structures [
258], neuroinflammation [
259,
260,
261,
262,
263], as well as central IR, which is also observed in patients with AD [
258,
262,
264]. Impaired insulin signaling may affect only certain areas of the brain, which is due to region-specific changes in the expression, distribution, and activity of INSR and downstream components of the insulin signaling system [
37,
265,
266,
267]. As in the case of T2DM, patients with T1DM have impaired CNS function, which is referred to as “diabetic encephalopathy”. Its characteristic features are cerebral atrophy, lesions of the white matter of the brain, impaired synaptic plasticity, delayed latency of evoked potentials, as well as a decrease in the activity of a number of cognitive domains. The main causes of diabetic encephalopathy are acute insulin deficiency, severe hyperglycemia, hyperactivation of oxidative and inflammatory processes in the brain, as well as impaired blood circulation in the brain vessels, which differs in etiology and pathogenesis from that in T2DM [
268].
In T2DM and MS, due to IR, the receptor-mediated transport of insulin across the BBB is weakened, which can lead to insulin deficiency in brain structures, despite normal or elevated insulin levels in the blood. A certain contribution to the disruption of such transport is made by the disintegration of the BBB, which occurs in conditions of DM2 and MS as a result of the activation of inflammatory and apoptotic processes, an increase in the production of reactive oxygen species, and a decrease in the sensitivity of the cells that form the BBB to insulin [
65,
269]. Therefore, under conditions of peripheral IR in T2DM, the weakening of insulin signaling in the brain, or at least in certain brain regions, may be due not only to central IR but also to the insufficient transfer of insulin from the blood to brain. We have shown that in rats and mice with hyperinsulinemia and severe IR, the level of insulin in the hypothalamus and other brain regions is reduced, which indicates the development of insulin deficiency in the CNS under conditions of systemic hyperinsulinemia [
270,
271]. Proteolytic enzymes, including the insulin-degrading enzyme, capable of cleaving the insulin molecule, can make a certain contribution to the regulation of insulin levels in the brain. At low insulin concentrations, the insulin-degrading enzyme hydrolyzes β-amyloid peptides with high efficiency, thereby preventing the formation of β-amyloid plaques and the AD development. However, under conditions of hyperinsulinemia in T2DM and MS, this enzyme begins to hydrolyze insulin, which, competing with β-amyloid peptides, promotes the accumulation of β-amyloid aggregates in cerebral vessels, which is the reason for the close relationship between T2DM and AD [
272,
273,
274,
275].
At present, studies of the effect of INI on functional disorders in DM can be divided into two groups. The first, most extensive group consists of experimental and clinical studies to study the restorative effect of INI on the functional activity of different brain regions, neuronal networks, and cognitive functions, and significant progress has been made in this regard. The second, relatively small group consists of works devoted to the ability of INI to improve hormonal and metabolic parameters in DM and restore the functions of peripheral organs and tissues [
29,
36,
276].
The emphasis in the use of INI in patients with T2DM on the correction of cognitive deficit and the prevention of neurodegenerative changes is largely due to the fact that there is strong evidence that T2DM and AD are closely interconnected [
277,
278]. Thus, it has been established that the most important factors for the development of AD are prolonged hyperglycemia, decreased insulin sensitivity, increased inflammatory processes, including the development of neuroinflammation, and lipotoxicity, which are typical for patients with T2DM [
278]. Since INI is widely used for the treatment of AD, it was assumed that insulin was highly effective in the treatment of elderly patients with T2DM and MS with signs of CNS damage, which was subsequently confirmed by clinical studies [
29,
279,
280,
281]. Importantly, both patients with T2DM have an increased risk of developing AD, and patients with AD and vascular dementia have a predisposition to T2DM [
274,
275,
282]. That is, the etiology and pathogenesis of T2DM and AD are a two-way avenue. Thus, the treatment of one of these pathologies, including with the use of INI, can largely prevent the development of the other.
6. Intranasal Insulin and Anesthesia
Cognitive disorders are one of the most common postoperative complications. Postoperative delirium (POD) develops as early as 5–7 days after surgery, which takes place with the use of anesthesia. Later, POD leads to cognitive impairment, which significantly increases the duration of treatment and hospitalization, worsens the prognosis of surgical intervention, and increases its mortality [
315]. Despite the fact that the pathophysiology of POD is not well understood, it is believed that its main mechanisms are impaired neuroendocrine regulation, neuroinflammation, neural network dysconnectivity, as well as ischemic damage caused by a decrease in CBF [
316]. The consequence of postoperative ischemia is severe metabolic disorders in the brain tissue due to the inhibition of aerobic processes, which is illustrated by an increase in the level of lactate in the cerebrospinal fluid and a decrease in the activity of neuron-specific enolase [
315,
317,
318,
319].
Cognitive disorders that develop after POD are divided into delayed neurocognitive recovery (DNR), which is due to neurocognitive impairment observed in patients from the 7th to the 30th day after surgery, and postoperative neurocognitive disorder (pNCD), which is observed from the 30th day to one year after surgery [
320]. The frequency of DNR and pNCD depends on many factors, such as age; type of surgical intervention; comorbidities (DM, respiratory diseases, etc.); psychophysiological state of the patient; and perioperative hyperglycemia, as well as tests used to assess cognitive disorders [
320,
321,
322,
323]. For the development of DNR and pNCD, the type and depth of anesthesia are important, and among the pathophysiological mechanisms that determine the relationship between postoperative cognitive disorders and anesthesia, it is necessary to highlight mitochondrial dysfunctions, increased inflammatory and apoptotic processes in neurons, hyperphosphorylation of the tau protein, and also the accumulation of β-amyloid proteins and the formation of their aggregates [
324,
325,
326,
327].
7. Intranasal Insulin and the Gonadal and Thyroid Systems
In addition to controlling metabolic processes and functioning as a neuroprotector, insulin, such as other peptides of the insulin group, plays an important role in the regulation of the functioning of the neuroendocrine and endocrine systems. This is due to the fact that both peripheral and central links of the hypothalamic-pituitary system are targets for insulin, since they contain all the main components of the insulin signaling system [
347,
348].