A decrease in the activity of the insulin signaling system of the brain leads to neurodegeneration and impaired regulation of appetite, metabolism, and endocrine functions. This is due to the neuroprotective properties of brain insulin, its leading role in maintaining glucose homeostasis in the brain, as well as in the regulation of the brain signaling network responsible for the functioning of the nervous, endocrine and other systems. One of the approaches to restore the brain insulin system is the use of intranasally administered insulin (INI). Currently, INI is being considered as a promising drug to treat Alzheimer's disease and mild cognitive impairment. Clinical application of INI is being developed for the treatment of other neurodegenerative diseases and improve cognitive abilities in stress, overwork, and depression. In recent years, it has been shown that INI can be used for the treatment of cerebral ischemia, traumatic brain injuries, postoperative delirium, as well as diabetes mellitus and its complications, including dysfunctions in the gonadal and thyroid axes.
- intranasal insulin
- brain
- diabetes mellitus
- cerebral ischemia
- postoperative delirium
- traumatic brain injury
- gonadal axis
- thyroid axis
- neurodegeneration
1. Introduction
2. Insulin and Insulin Signaling System in the Brain and Intranasal Delivery of Insulin to the Brain
2.1. Sources of Insulin in the Brain
The origin of insulin in the brain is associated mainly with the intake of hormone produced by β-cells from the bloodstream due to its transport through the blood–brain barrier (BBB). However, a high level of insulin in the brain during the neonatal period of development, when the insulin-producing function of the pancreas is either absent or still insufficient for the synthesis of the required amounts of the hormone, indicates the possibility of insulin synthesis in the brain de novo, at least at the early stages of ontogeny. This is evidenced by the presence of mRNA for proinsulin in a number of brain regions in rat embryos and in newborn rats and rabbits [50], as well as in the culture of neurons obtained from various regions of the rabbit embryonic brain [51]. In this regard, it is important to note that neurons, such as pancreatic β-cells, are electrically excitable and respond by depolarization and exocytosis to the action of hormones and to an increase in glucose levels, which is very important for the synthesis and secretion of insulin by them, since these processes require depolarization of ATP-sensitive potassium channels functionally active in both cell types [52,53][52][53]. The entry of insulin circulating in the blood into the brain through the BBB is carried out using three main mechanisms. The first includes the transport of insulin to brain structures using receptor-mediated endocytosis (transcytosis), which is based on the binding of the hormone to insulin receptors (INSRs) located on the surface of endothelial cells, the subsequent internalization of insulin-receptor complexes included in vesicles into cytoplasm, and their transfer to the opposite side of the endothelial cell, followed by exocytosis on its abluminal side and the release of insulin towards the endings of neurons or glial cells [54,55][54][55]. The decisive role of INSR in this process is evidenced by the fact that in mice with a knockout of the Insr gene in endothelial cells, the permeability of the BBB for insulin deteriorates sharply, and as a result, insulin signaling in the hypothalamus, hippocampus, and prefrontal cortex is impaired [56]. There is competition between insulin and insulin-like growth factor-1 (IGF-1), another member of the insulin family, for receptors that mediate the transport of insulin across the BBB, since IGF-1 at higher than physiological concentrations is able to suppress insulin transcytosis [57]. Insulin transcytosis is regulated by hormones, lipids, and vasodilation factors. It significantly depends on the physiological parameters, weakens with obesity and MS, and increases with inflammatory diseases and insulin-deficient forms of DM, although the mechanisms in each case differ and are not always investigated [55,58,59,60,61,62,63,64,65][55][58][59][60][61][62][63][64][65]. Astrocytes and pericytes, which form close contacts with vascular endothelial cells, play an important role in modulating transendothelial insulin transport across the BBB [66]. The loss of pericytes or a violation of their structure leads to an increase in the permeability of the BBB and an impaired selectivity of transport of insulin and other hormones through the BBB [67,68,69][67][68][69]. Other mechanisms include passive insulin transport without INSR involvement through the fenestrated capillaries and ependymal cells of the median eminence, located below the mediobasal hypothalamus, ventral to the third ventricle and adjacent to the arcuate nucleus of the hypothalamus [70]. The transport of insulin across the median eminence is regulated by tanycytes, highly specialized ependymal cells that line the ventrolateral wall and the floor of the third ventricle and are involved in the exchange of substances between the blood and cerebrospinal fluid [71,72][71][72]. The presence of tight junctions between adjacent tanycytes provides a physical barrier that controls the transfer of insulin from portal capillaries and cerebrospinal fluid to the hypothalamus [73,74][73][74]. Remodeling of the hemato-hypothalamic barrier, for example, due to changes in blood glucose levels, affects the transfer of insulin and other circulating hormones through it to the arcuate nuclei of the hypothalamus [73,75][73][75]. In this regard, it should be noted that insulin enters the arcuate nuclei of the hypothalamus mainly through its passive transport with the participation of tanycytes, and not through receptor-mediated transcytosis [72].2.2. Insulin Signaling System
A key component of the insulin signaling system in brain neurons and glial cells are INSRs, which are found in the hypothalamus, hippocampus, cerebellum, thalamus, cerebral cortex, olfactory bulb, dentate gyrus, motor cortex, and other regions [76,77,78,79,80,81,82,83,84,85][76][77][78][79][80][81][82][83][84][85]. In mammals, two INSR isoforms are present, the full-length (INSR-B) and the truncated (INSR-A). INSR-A is formed as a result of alternative splicing of the 11th exon by removing a 12 amino acid segment from the encoded sequence. These INSR isoforms are co-expressed in many tissues, but the ratio of INSR-A/INSR-B in the brain is much higher than at the periphery, which is why INSR-A is often referred to as the neuronal isoform of the receptor [86,87,88][86][87][88]. INSR-A and INSR-B are similar in structural organization and pharmacological characteristics, although they have a number of functional differences, including those due to differences in glycosylation [89,90,91][89][90][91]. Insulin is also able, albeit with low affinity, to bind to a hybrid receptor that includes the monomers of both INSR and IGF-1 receptor (IGF1R). The IGF1R is structurally and functionally similar to INSR, since the genes encoding these receptors, Igf1r and Insr, originate from a common ancestral gene and are part of an ancient, highly conserved signaling system in vertebrates and invertebrates involved in the regulation of cell metabolism, growth, and differentiation [92,93][92][93]. Both INSR isoforms are α2β2-heterotetramers in which extracellular α-subunits and transmembrane β-subunits are linked by disulfide bonds [94,95][94][95]. The α-subunit and the N-terminal segment of the β-subunit form the INSR ectodomain responsible for high-affinity insulin binding. The large cytoplasmic domain of the β-subunit contains a highly conserved tyrosine kinase domain that is stimulated after insulin binds to the ligand-binding site located in the α-subunit [94]. Binding of the α-subunit to insulin results in conformational rearrangements that affect the tyrosine kinase domain of the β-subunit and induce receptor autophosphorylation at the tyrosine residues, Tyr1158, Tyr1162 and Tyr1163, the main targets of the receptor tyrosine kinase. After autophosphorylation, the β-subunit interacts with regulatory and adapter proteins that contain phosphotyrosine-binding sites that can specifically interact with segments including phosphorylated Tyr1158, Tyr1162 and Tyr1163 (Figure 1). Depending on the pattern of this interaction, various signaling pathways are activated, resulting in different responses of the target cell to insulin [96].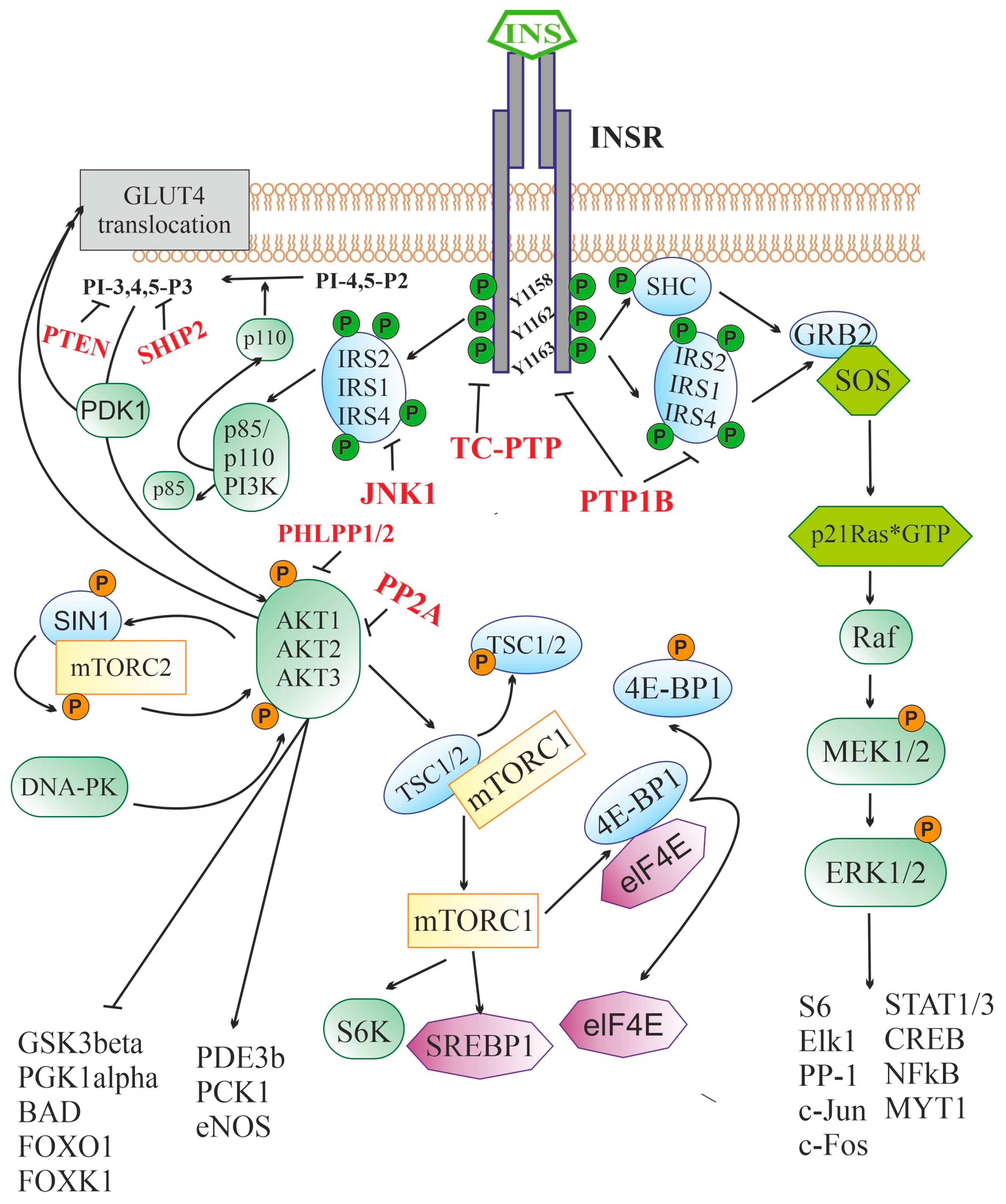
2.3. The Intranasal Route of Insulin Delivery
Over the past decades, the intranasal route of administration has become relevant for the delivery of drugs to the CNS, bypassing the BBB. More than 30 years ago, William H. Frey II was the first to propose the use of intranasal administration of various biologically active substances, including polypeptide hormones and growth factors, for the treatment of Alzheimer’s disease and other diseases of the nervous system, and insulin was of the greatest interest among the studied hormones [131,132,133][131][132][133]. To date, there are already more than 50 clinical trials of INI in both healthy subjects and patients with CNS diseases and diabetes (https://clinicaltrials.gov/, accessed on 27 December 2022). The main advantage of intranasal administration of drugs is the targeted delivery to the CNS of substances that are unable to penetrate the BBB due to their size, charge, or other physical, chemical and biological properties. This approach provides a rapid achievement of a therapeutic effect, as well as non-invasiveness and ease of use compared to oral or injection methods of drug delivery. An important feature of intranasal administration is the absence of passage through the hepatic barrier, which allows the use of lower doses of drugs that reduce side effects [16,20,21][16][20][21]. Significant progress in the study of the INI mechanisms involved in bypassing the BBB, its pharmacokinetics and pharmacodynamics, as well as the targets of INI in the brain, has been achieved through the use of animal models [16,17,18,19,134][16][17][18][19][134]. After nebulization in the nasal cavity, insulin enters the nasal mucosa, where it is transported to the brain by an intracellular route using receptor-mediated endocytosis via axons of olfactory and trigeminal nerves, which innervate the nasal cavities, as well as via extracellular pathway by interstitial fluid, but with different speeds. Retrograde axonal transport is rather slower [135] than an extracellular one, so it takes hours to reach the inner brain structures, and more likely that INI is primarily transported along extra-neuronal routes [16,136][16][136]. The extra-neuronal pathway rapidly allows insulin and its homolog IGF-1 to spread along the olfactory and trigeminal nerves and distribute within brain in the rostral to caudal direction [18,19,137][18][19][137]. Once in the CNS, insulin binds to brain INSRs and causes their activation, which is demonstrated by an increase in the level of tyrosine receptor phosphorylation [19]. This leads to activation of intracellular signaling pathways, including the PI-3-K/Akt signaling pathway, as mentioned above [17,18][17][18]. After 15–30 min after intranasal administration, insulin accumulation was shown in the olfactory bulbs, striatum, substantia nigra, brainstem, cerebellum, and, to a lesser extent, in the hippocampus and cerebral cortex [17,18][17][18]. According to data from other authors, large amounts of intranasally administered insulin were found in the cortex, cerebellum, hippocampus, and hypothalamus [19]. Thereafter, the amount of insulin in the brain gradually decreases but remains elevated even 6 h after its administration [18]. An increase in insulin levels after intranasal administration was also found in the cerebrospinal fluid and in a small amount in the serum [134,138,139][134][138][139]. With long-term (9 days) administration to mice, INI increased the level of glucose, adenosine triphosphate, and phosphocreatine in the brain of animals, which indicates its positive effect on energy metabolism in the brain [134]. In this regard, it should be noted that in humans, a positive effect of INI on improving glucose homeostasis and energy metabolism in the brain has been demonstrated, which is important for correcting hypometabolic conditions in neurodegenerative and other diseases [140]. In human studies, INI bypassed the BBB and detected in the cerebrospinal fluid at 10 min with peak at 30 min and remained significantly elevated at 80 min [24]. The serum insulin levels peaked in 10–20 min after its intranasal administration, then remained slightly elevated for approximately 1 h and returned to mean values 1.5–2 h after nebulization with an insignificant risk of hypoglycemia mainly in a fasting state [25,141][25][141]. The fast and unhindered entry of INI into the CNS is a great advantage of the intranasal route of hormone administration compared to other routes of its administration, which is important in pathologies that require emergency treatment, such as stroke and brain injury, as well as during pre- and postoperative procedures. The data obtained from the use of INI in humans does not raise any significant concerns in its short-term and long-term use. On the contrary, most studies point to its positive effect on cognitive functions, good tolerability, and the absence of serious side effects that could become an obstacle to INI therapy. However, it should be noted that the chronic, long-term use of INI in a small cohort of patients still revealed some undesirable effects, which are mainly due to the procedure of spraying INI in the nasal cavity (rhinitis, mild epistaxis, sneezing, headaches, and predisposition to respiratory tract infections) [26,27,28,33,46][26][27][28][33][46]. It is important that these undesirable effects can be eliminated by optimizing both the procedure for intranasal nebulization of the hormone and the composition of the mixture for nebulization, since they are not caused by the toxic effect of INI on the nasal cavity and brain structures.3. Intranasal Insulin and Brain Ischemia
Cerebral ischemia occurs as a result of various critical events and diseases, including surgery, resuscitation, traumatic brain injury, asphyxia, hemorrhages, embolisms, myocardial infarction, acute blood loss, and hypotension [142,143,144][142][143][144]. However, the main cause of damage to brain neurons is a deficiency of oxygen and nutrients in the CNS. The consequences of cerebral ischemia are determined by the level of damage and death of neurons and glial cells and depend on the degree of deterioration of blood flow in the brain tissue, the duration of the ischemic episode, and the area of the ischemic lesion. The metabolic and hormonal status of the brain and organism before cerebral ischemia is also of great importance for the severity of CNS damage caused by it [143]. Developing glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, inhibition of protein synthesis, and activation of proteolytic enzymes lead to the triggering of endoplasmic reticulum stress, cell death, and neuroinflammation. Prolonged brain ischemia leads to disintegration and irreversible changes in the brain signaling cascades responsible for the survival and functional activity of neurons, resulting in their death through apoptosis, necrosis, or autophagy [145]. Not only neurons but also glial cells are damaged during ischemia, which negatively affects the neuroprotective potential of glia. At the brain level, cerebral ischemia is accompanied by cerebral edema, damage to the integrity and permeability of the BBB, hyperactivation of microglia, and increased pro-inflammatory cascades [146,147,148,149][146][147][148][149]. During ischemia, compensatory mechanisms are activated that maintain the viability of neurons and reduce the intensity of pro-inflammatory and apoptotic processes in the CNS. These mechanisms include signaling cascades triggered by a large number of neurohormones, growth factors and cytokines with neuroprotective properties, and the insulin and IGF-1 signaling cascades are of key importance among them [145]. As a result, the use of hormonal regulators with neuroprotective properties, including insulin, is able to compensate for ischemic brain damage, which makes them promising drugs for the treatment of ischemia. Despite many years of research in the field of treatment of ischemic stroke, there are currently no anti-ischemic neuroprotective drugs that are used clinically [150,151][150][151]. Since the pathophysiology of stroke is multi-faceted, it is promising to use neuroprotectors with a pleiotropic effect, which makes it possible to prevent several pathological processes in the brain at once. This predetermines the main direction of the search for pharmacological agents to treat postischemic dysfunctions of the nervous and other systems. Hyperglycemia leads to more severe consequences [152,153,154][152][153][154], making glycemic control one of the most important steps in the treatment and rehabilitation of patients with cerebral ischemia [155]. In this regard, it should be noted that both hyperglycemia and hypoglycemia, including those induced by insulin therapy, are associated with a poor prognosis of ischemia [156,157][156][157]. In this regard, it is not surprising that patients with DM or pre-diabetes have an increased risk of ischemic and hemorrhagic stroke [158,159,160,161][158][159][160][161]. Along with this, about half of the patients who had an ischemic stroke later had impaired glucose homeostasis and IR [162,163][162][163]. T2DM increases the risk of recurrent stroke after the first episode of ischemia or stroke, so secondary prevention of stroke in patients with metabolic disorders is also an important problem [150,151][150][151]. There is evidence that weight loss, restoration of tissue sensitivity to insulin, and normalization of glucose homeostasis are very effective in stroke prevention [150,151,158,159,160,161][150][151][158][159][160][161]. All of the above indicates that injectable forms of insulin, which normalizes glucose levels in hyperglycemia, can be indicated for the correction of hyperglycemic conditions in diabetic patients with stroke, as well as in non-diabetic patients with impaired glucose homeostasis that developed as a result of a stroke. However, in the recent years, the focus of the use of insulin has shifted from its metabolic effects to neuroprotective effect and the ability of insulin to influence the functional activity of brain structures that have undergone ischemic damage. In connection with the foregoing, let us consider the currently available data on the effect of insulin and its functional homolog, IGF-1, on postischemic damage in animals with experimental ischemic and hemorrhagic stroke and hypoxia-ischemia. The neuroprotective properties of insulin in ischemic injury have been studied for more than 30 years [164,165,166,167][164][165][166][167]. It has been shown that insulin with various delivery methods (subcutaneous, intraperitoneal, intravenous, and intracerebroventricular) positively affects the survival and cognitive functions of animals that have undergone cerebral ischemia [165,168,169,170,171,172,173,174,175,176][165][168][169][170][171][172][173][174][175][176]. The antiapoptotic signaling pathway, including IR/IRS/PI3K/AKT, plays a leading role in the implementation of the neuroprotective action of insulin. By activating AKT, insulin suppresses the release of cytochrome C from mitochondria, thereby preventing the translocation of the proapoptotic protein BAX to them [177[177][178],178], and also stimulates synthesis of neuroprotective proteins, including the antioxidant enzymes [179[179][180],180], which increases the survival of neurons and prevents cognitive dysfunctions in animals with cerebral ischemia [178]. The contribution of insulin to the improvement of cognitive functions during oxidative stress or excitotoxicity may also be due to its neuromodulatory effect, by changing the surface density and activity of the glutamate and γ-aminobutyric acid ionotropic receptors in neurons, as well as by regulating the membrane transport of glutamate and γ-aminobutyric acid, which allows optimizing their ratio in the extracellular space [180]. The vasoactive effect of insulin is widely known, which, along with its neuroprotective effect, plays an important role in the implementation of the protective functions of insulin in cerebral ischemia. AKT-dependent activation of endothelial NO-synthase under the action of insulin on the endothelium of cerebral vessels leads to changes in the vascular lumen and improves the cerebral blood flow (CBF) [181,182][181][182]. At the same time, depending on the dose of the hormone and the functional state of the CNS, insulin is able to both increase and decrease the lumen of the vessels, exerting a modulating effect on CBF [183,184][183][184]. In healthy subjects, an increase in insulin levels in the brain leads to increased CBF and improved blood supply to brain tissue [185]. Despite a significant number of studies on the effects of peripheral and intracerebroventricular insulin administration to correct the consequences of cerebral ischemia, there are relatively few data on the use of INI, despite the promise of this method for correcting ischemic damage [186,187,188,189,190,191,192][186][187][188][189][190][191][192]. It should be noted that intranasally administered IGF-1 has been used for the correction of damage caused by cerebral ischemia–reperfusion for 20 years [186,193,194,195,196,197,198,199][186][193][194][195][196][197][198][199]. The targets of action of insulin and IGF-1 in the CNS largely coincide, which is due to both the similar architecture of their signaling cascades and the ability of insulin and IGF-1 to activate the same receptors, including hybrid di/oligomeric forms of INSR/IGF1R [95]. In addition, both hormones have a similar pattern of regulatory effects on neurons and glial cells, exerting antiapoptotic and anti-inflammatory effects, as demonstrated for insulin in the use of INI for the treatment of neurodegenerative diseases, including AD [27,200,201][27][200][201]. All this allowed the group of Vera Novak to put forward a hypothesis about the prospects of using INI to correct cerebral ischemia [186]. It is important that INI has clear advantages over intranasally administered IGF-1, because, unlike insulin, IGF-1 is characterized by a very powerful mitogenic effect, which has significant risks of cell malignancy, and the IGF-1 dosage form is expensive and has very limited clinical experience. It is also important that INI not only has a neuroprotective effect on neurons and glial cells but also improves the brain blood supply, which is especially valuable in the conditions of cerebral ischemia [185]. The potential mechanisms of neuroprotective action of INI in cerebral ischemia are shown in Figure 2.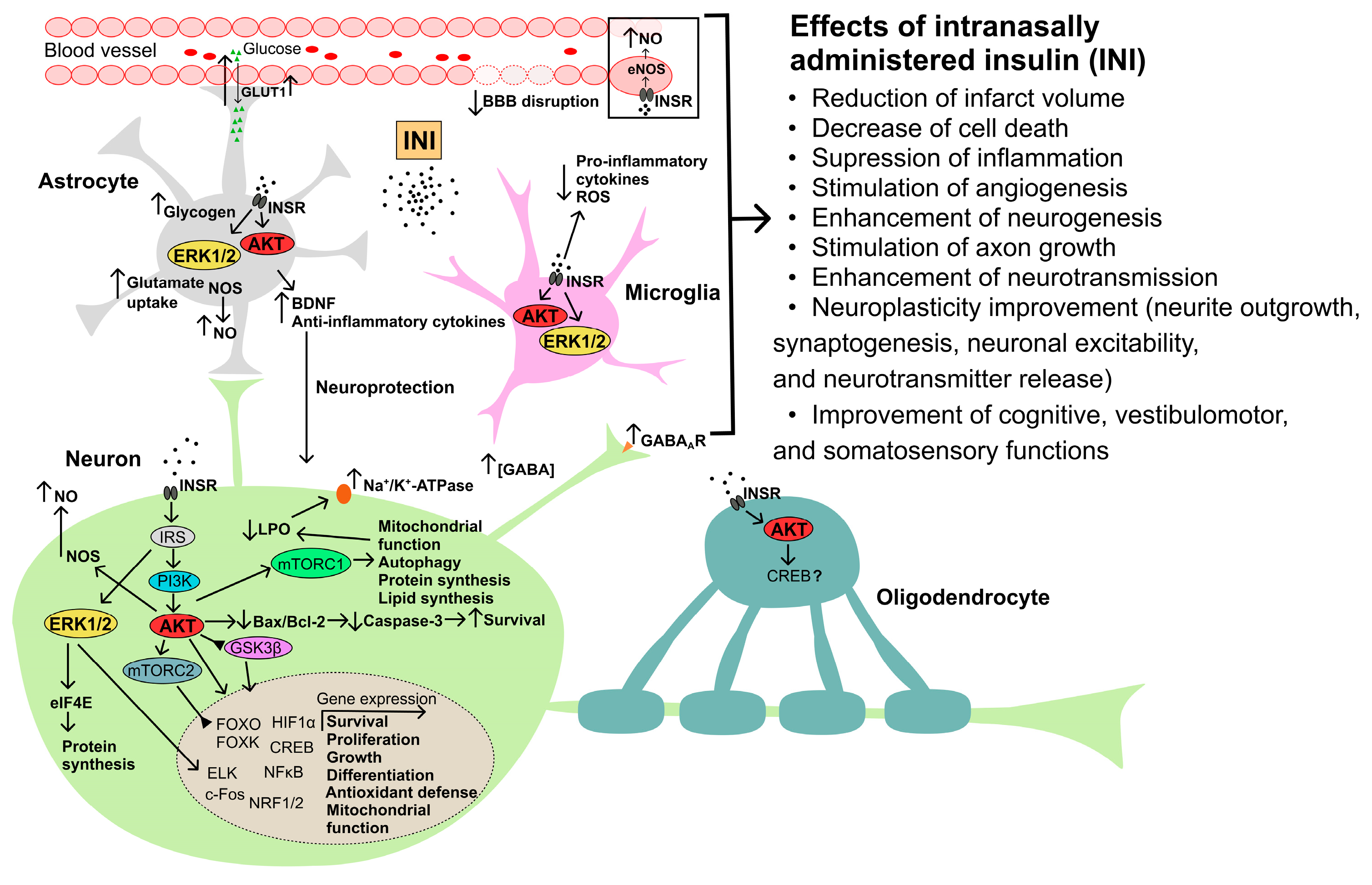
4. Intranasal Insulin and Brain Injury
Traumatic brain injury (TBI) in most cases leads to long-term cognitive deficit, which is currently poorly or not treatable at all [238][202]. Even mild TBIs in 15% of patients lead to cognitive deficits in the long term [239][203]. TBI is based on a three-phase response of the brain to an external damaging effect [240][204]. The first phase corresponds to the development immediately after injury of a hypermetabolic state caused by a powerful release of ions and glutamate, which is due to a sharply increased demand for energy. The second phase corresponds to a significant decrease in the absorption of glucose by the brain tissue, which leads to an energy deficit in neurons and is the main cause of brain damage and triggering neurodegenerative processes. The long-term consequences of TBI, including the subsequently developing cognitive deficit, largely depend on the duration and severity of this phase. The final phase includes a gradual return of glucose uptake by the brain to normal values [240][204]. It is important that the restoration of metabolism can occur only in some regions of the brain, while other regions are characterized by a decrease in metabolic status over a long period, years after TBI, even in patients with a mild form of this injury [241,242][205][206]. The disintegration of functional connections between various brain structures, the decrease in neuronal plasticity in them, and the activation of neuro-inflammatory processes lead to dysfunctions of the CNS and an imbalance of metabolic processes at the periphery [243,244,245][207][208][209]. In animals with experimental TBI, a decrease in brain sensitivity to insulin was found, which may be a consequence of glutamate excitotoxicity induced by traumatic exposure and activation of pro-inflammatory processes in the brain [244,246,247][208][210][211]. With a single traumatic impact, a decrease in the brain sensitivity to insulin is observed within 7 days, and with repeated trauma, up to 28 days [246][210]. At the same time, in obese mice, which initially had impaired insulin signaling in the brain, the consequences of TBI were expressed to a much greater extent. They were accompanied by a significant weakening of the stimulatory effect of insulin on the INSR/IRS/PI3K/AKT signaling pathway, pronounced neuroinflammation, a significant deterioration in learning ability, a sharp decrease in memory, and a severe anxiety-depressive state [246][210]. A study of glucose uptake in the brain of rats showed that after a mild TBI, its changes begin to be registered as early as 3–6 h after the traumatic impact and persist to some extent for 5–10 days, and this effect was specific both for certain regions of the brain and for certain types of neurons and glial cells [248,249,250][212][213][214]. The development of post-traumatic complications is largely due to insulin sensitivity of the brain, which decreases with obesity and a number of other disorders, as well as the effectiveness of integrative interactions between brain regions, which can significantly affect insulin sensitivity and glucose uptake by neurons and glial cells [244,250][208][214]. Since after a single TBI, there is a transient weakening of the sensitivity of certain regions of the brain to insulin, it seems quite logical that repeated TBIs aggravate the central IR, especially when they occur in the time window of a decrease in insulin sensitivity caused by previous TBI. In 2022, a model of lateral fluid-percussion injury in rats showed that 4 months after traumatic exposure, glucose uptake was reduced in the hippocampus, ipsilateral striatum, and frontoparietal cortex [251][215]. Decreased glucose uptake in the stratum was positively correlated with deterioration in spatial memory [251][215]. All of the above points to the potential ability of INI to improve glucose homeostasis in the brain and compensate, at least in part, for the hypometabolic state that develops after TBI [244][208]. This is supported by the data on INI effect on adult male Sprague Dawley rats with moderate controlled cortical injury. Treatment of animals with INI (Humulin R-100, 6 IU/rat) was started 4 h after traumatic exposure, and then INI treatment at the same dose was continued for two weeks [252][216]. As a result, glucose utilization was normalized in the hippocampus of rats treated with INI, which was accompanied by a decrease in the hippocampal lesion area, a decrease in neuroinflammation mediated by microglia, and an improvement in memory and spatial orientation [252][216]. The prospects for the use of INI in TBI are also indirectly evidenced by the fact that moderate and severe TBI is significantly associated with AD, which is one of the targets of INI [253][217]. The severity of brain damage is positively correlated with the incidence of AD. It should be noted that a close relationship is also observed between AD and Parkinson’s disease, which have many common molecular causes, including metabolic changes in the brain that provoke neurodegeneration, and both are therapeutic targets for INI [254][218]. It should also be noted that the pathogenetic mechanisms of stroke and TBI are quite similar, and therefore the previously described neuroprotective effects of INI for cerebral ischemia can also be manifested in TBI [255][219]. The possible mechanisms and targets of neuroprotective action of INI in TBI are summarized in Figure 2. Thus, the general pathogenetic factors of CNS diseases may a priori suggest the potential effectiveness of INI for the treatment of TBI and the prevention of its negative consequences.5. Intranasal Insulin and Diabetes Mellitus
DM is characterized by significant changes in the functional activity of the insulin signaling system, and this is due to both absolute or relative insulin deficiency in type 1 DM (T1DM) and the development of IR in T2DM. In both cases, the result is a weakening of insulin signaling pathways both in the brain and at the periphery. To restore them, pharmacological approaches can be used to compensate for insulin deficiency (insulin replacement therapy) or to reduce IR (metformin and other drugs that increase insulin sensitivity). Since insulin in the brain is involved in the regulation of the functions of neurons and glial cells, controls the integrative relationships between different brain regions, and mediates the central regulation of physiological and metabolic processes at the periphery, an impaired brain insulin signaling in DM leads to a wide range of disorders. As a result, the use of INI, which activates the insulin system in the brain, may be useful for preventing cognitive deficits in patients with DM, as well as for restoring the central insulin-mediated regulation of peripheral metabolism and physiological functions. Among the factors that lead to the development of neurodegenerative processes and cognitive deficit in T2DM and MS, the most important are cerebrovascular dysfunctions [256[220][221],257], reduced glucose metabolism in brain structures [258][222], neuroinflammation [259[223][224][225][226][227],260,261,262,263], as well as central IR, which is also observed in patients with AD [258,262,264][222][226][228]. Impaired insulin signaling may affect only certain areas of the brain, which is due to region-specific changes in the expression, distribution, and activity of INSR and downstream components of the insulin signaling system [37,265,266,267][37][229][230][231]. As in the case of T2DM, patients with T1DM have impaired CNS function, which is referred to as “diabetic encephalopathy”. Its characteristic features are cerebral atrophy, lesions of the white matter of the brain, impaired synaptic plasticity, delayed latency of evoked potentials, as well as a decrease in the activity of a number of cognitive domains. The main causes of diabetic encephalopathy are acute insulin deficiency, severe hyperglycemia, hyperactivation of oxidative and inflammatory processes in the brain, as well as impaired blood circulation in the brain vessels, which differs in etiology and pathogenesis from that in T2DM [268][232]. In T2DM and MS, due to IR, the receptor-mediated transport of insulin across the BBB is weakened, which can lead to insulin deficiency in brain structures, despite normal or elevated insulin levels in the blood. A certain contribution to the disruption of such transport is made by the disintegration of the BBB, which occurs in conditions of DM2 and MS as a result of the activation of inflammatory and apoptotic processes, an increase in the production of reactive oxygen species, and a decrease in the sensitivity of the cells that form the BBB to insulin [65,269][65][233]. Therefore, under conditions of peripheral IR in T2DM, the weakening of insulin signaling in the brain, or at least in certain brain regions, may be due not only to central IR but also to the insufficient transfer of insulin from the blood to brain. WeIt haves been shown that in rats and mice with hyperinsulinemia and severe IR, the level of insulin in the hypothalamus and other brain regions is reduced, which indicates the development of insulin deficiency in the CNS under conditions of systemic hyperinsulinemia [270,271][234][235]. Proteolytic enzymes, including the insulin-degrading enzyme, capable of cleaving the insulin molecule, can make a certain contribution to the regulation of insulin levels in the brain. At low insulin concentrations, the insulin-degrading enzyme hydrolyzes β-amyloid peptides with high efficiency, thereby preventing the formation of β-amyloid plaques and the AD development. However, under conditions of hyperinsulinemia in T2DM and MS, this enzyme begins to hydrolyze insulin, which, competing with β-amyloid peptides, promotes the accumulation of β-amyloid aggregates in cerebral vessels, which is the reason for the close relationship between T2DM and AD [272,273,274,275][236][237][238][239]. At present, studies of the effect of INI on functional disorders in DM can be divided into two groups. The first, most extensive group consists of experimental and clinical studies to study the restorative effect of INI on the functional activity of different brain regions, neuronal networks, and cognitive functions, and significant progress has been made in this regard. The second, relatively small group consists of works devoted to the ability of INI to improve hormonal and metabolic parameters in DM and restore the functions of peripheral organs and tissues [29,36,276][29][36][240]. The emphasis in the use of INI in patients with T2DM on the correction of cognitive deficit and the prevention of neurodegenerative changes is largely due to the fact that there is strong evidence that T2DM and AD are closely interconnected [277,278][241][242]. Thus, it has been established that the most important factors for the development of AD are prolonged hyperglycemia, decreased insulin sensitivity, increased inflammatory processes, including the development of neuroinflammation, and lipotoxicity, which are typical for patients with T2DM [278][242]. Since INI is widely used for the treatment of AD, it was assumed that insulin was highly effective in the treatment of elderly patients with T2DM and MS with signs of CNS damage, which was subsequently confirmed by clinical studies [29,279,280,281][29][243][244][245]. Importantly, both patients with T2DM have an increased risk of developing AD, and patients with AD and vascular dementia have a predisposition to T2DM [274,275,282][238][239][246]. That is, the etiology and pathogenesis of T2DM and AD are a two-way avenue. Thus, the treatment of one of these pathologies, including with the use of INI, can largely prevent the development of the other.6. Intranasal Insulin and Anesthesia
Cognitive disorders are one of the most common postoperative complications. Postoperative delirium (POD) develops as early as 5–7 days after surgery, which takes place with the use of anesthesia. Later, POD leads to cognitive impairment, which significantly increases the duration of treatment and hospitalization, worsens the prognosis of surgical intervention, and increases its mortality [315][247]. Despite the fact that the pathophysiology of POD is not well understood, it is believed that its main mechanisms are impaired neuroendocrine regulation, neuroinflammation, neural network dysconnectivity, as well as ischemic damage caused by a decrease in CBF [316][248]. The consequence of postoperative ischemia is severe metabolic disorders in the brain tissue due to the inhibition of aerobic processes, which is illustrated by an increase in the level of lactate in the cerebrospinal fluid and a decrease in the activity of neuron-specific enolase [315,317,318,319][247][249][250][251]. Cognitive disorders that develop after POD are divided into delayed neurocognitive recovery (DNR), which is due to neurocognitive impairment observed in patients from the 7th to the 30th day after surgery, and postoperative neurocognitive disorder (pNCD), which is observed from the 30th day to one year after surgery [320][252]. The frequency of DNR and pNCD depends on many factors, such as age; type of surgical intervention; comorbidities (DM, respiratory diseases, etc.); psychophysiological state of the patient; and perioperative hyperglycemia, as well as tests used to assess cognitive disorders [320,321,322,323][252][253][254][255]. For the development of DNR and pNCD, the type and depth of anesthesia are important, and among the pathophysiological mechanisms that determine the relationship between postoperative cognitive disorders and anesthesia, it is necessary to highlight mitochondrial dysfunctions, increased inflammatory and apoptotic processes in neurons, hyperphosphorylation of the tau protein, and also the accumulation of β-amyloid proteins and the formation of their aggregates [324,325,326,327][256][257][258][259].7. Intranasal Insulin and the Gonadal and Thyroid Systems
In addition to controlling metabolic processes and functioning as a neuroprotector, insulin, such as other peptides of the insulin group, plays an important role in the regulation of the functioning of the neuroendocrine and endocrine systems. This is due to the fact that both peripheral and central links of the hypothalamic-pituitary system are targets for insulin, since they contain all the main components of the insulin signaling system [347,348][260][261].References
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829.
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741.
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimer′s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960.
- Cai, Z.; Xiao, M.; Chang, L.; Yan, L.J. Role of insulin resistance in Alzheimer’s disease. Metab. Brain Dis. 2015, 30, 839–851.
- De la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimers Dis. 2018, 62, 1381–1390.
- De la Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83.
- Eraky, S.M.; Ramadan, N.M.; Abo El-Magd, N.F. Ameliorative effects of bromelain on aluminum-induced Alzheimer’s disease in rats through modulation of TXNIP pathway. Int. J. Biol. Macromol. 2022, 227, 1119–1131.
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43.
- Ardanaz, C.G.; Ramírez, M.J.; Solas, M. Brain Metabolic Alterations in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3785.
- González, A.; Calfío, C.; Churruca, M.; Maccioni, R.B. Glucose metabolism and AD: Evidence for a potential diabetes type 3. Alzheimers Res. Ther. 2022, 14, 56.
- Neumann, K.F.; Rojo, L.; Navarrete, L.P.; Farías, G.; Reyes, P.; Maccioni, R.B. Insulin resistance and Alzheimer’s disease: Molecular links & clinical implications. Curr. Alzheimer Res. 2008, 5, 438–447.
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417.
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47.
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814.
- Ortiz, G.G.; Huerta, M.; González-Usigli, H.A.; Torres-Sánchez, E.D.; Delgado-Lara, D.L.; Pacheco-Moisés, F.P.; Mireles-Ramírez, M.A.; Torres-Mendoza, B.M.; Moreno-Cih, R.I.; Velázquez-Brizuela, I.E. Cognitive disorder and dementia in type 2 diabetes mellitus. World J. Diabetes 2022, 13, 319–337.
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52.
- Picone, P.; Sabatino, M.A.; Ditta, L.A.; Amato, A.; San Biagio, P.L.; Mulè, F.; Giacomazza, D.; Dispenza, C.; Di Carlo, M. Nose-to-brain delivery of insulin enhanced by a nanogel carrier. J. Control. Release 2018, 270, 23–36.
- Fan, L.W.; Carter, K.; Bhatt, A.; Pang, Y. Rapid transport of insulin to the brain following intranasal administration in rats. Neural Regen. Res. 2019, 14, 1046–1051.
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci. Rep. 2019, 9, 2621.
- Patel, D.; Patel, B.; Wairkar, S. Intranasal delivery of biotechnology-based therapeutics. Drug Discov. Today 2022, 27, 103371.
- Sharma, M.; Waghela, S.; Mhatre, R.; Saraogi, G.K. A Recent Update on Intranasal Delivery of High Molecular Weight Proteins, Peptides, and Hormones. Curr. Pharm. Des. 2021, 27, 4279–4299.
- Bose, M.; Farias Quipildor, G.; Ehrlich, M.E.; Salton, S.R. Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development. Cells 2022, 11, 3629.
- Duong, V.A.; Nguyen, T.T.; Maeng, H.J. Recent Advances in Intranasal Liposomes for Drug, Gene, and Vaccine Delivery. Pharmaceutics 2023, 15, 207.
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516.
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458.
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38.
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334.
- Novak, P.; Pimentel Maldonado, D.A.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364.
- Novak, V.; Mantzoros, C.S.; Novak, P.; McGlinchey, R.; Dai, W.; Lioutas, V.; Buss, S.; Fortier, C.B.; Khan, F.; Aponte Becerra, L.; et al. MemAID: Memory advancement with intranasal insulin vs. placebo in type 2 diabetes and control participants: A randomized clinical trial. J. Neurol. 2022, 269, 4817–4835.
- Benedict, C.; Frey, W.H., 2nd; Schiöth, H.B.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 2011, 46, 112–115.
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906.
- Ribarič, S. The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules 2016, 21, 689.
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510.
- Chen, J.; Hu, L.; Yang, G.; Hu, Q. Current Therapeutic Strategy in the Nasal Delivery of Insulin: Recent Advances and Future Directions. Curr. Pharm. Biotechnol. 2018, 19, 400–415.
- Muñoz-Jiménez, M.; Zaarkti, A.; García-Arnés, J.A.; García-Casares, N. Antidiabetic Drugs in Alzheimer’s Disease and Mild Cognitive Impairment: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2020, 49, 423–434.
- Gaddam, M.; Singh, A.; Jain, N.; Avanthika, C.; Jhaveri, S.; De la Hoz, I.; Sanka, S.; Goli, S.R. A Comprehensive Review of Intranasal Insulin and Its Effect on the Cognitive Function of Diabetics. Cureus 2021, 13, e17219.
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37.
- Hallschmid, M. Intranasal insulin. J. Neuroendocrinol. 2021, 33, e12934.
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2021, 8, 240–248.
- Miziak, B.; Błaszczyk, B.; Czuczwar, S.J. Some Candidate Drugs for Pharmacotherapy of Alzheimer’s Disease. Pharmaceuticals 2021, 14, 458.
- Long, C.; Han, X.; Yang, Y.; Li, T.; Zhou, Q.; Chen, Q. Efficacy of intranasal insulin in improving cognition in mild cognitive impairment or dementia: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 963933.
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334.
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans: Superiority of insulin aspart. Neuropsychopharmacology 2007, 32, 239–243.
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344.
- Kupila, A.; Sipilä, J.; Keskinen, P.; Simell, T.; Knip, M.; Pulkki, K.; Simell, O. Intranasally administered insulin intended for prevention of type 1 diabetes--a safety study in healthy adults. Diabetes Metab. Res. Rev. 2003, 19, 415–420.
- Schmid, V.; Kullmann, S.; Gfrörer, W.; Hund, V.; Hallschmid, M.; Lipp, H.P.; Häring, H.U.; Preissl, H.; Fritsche, A.; Heni, M. Safety of intranasal human insulin: A review. Diabetes Obes. Metab. 2018, 20, 1563–1577.
- Gwizdala, K.L.; Ferguson, D.P.; Kovan, J.; Novak, V.; Pontifex, M.B. Placebo controlled phase II clinical trial: Safety and efficacy of combining intranasal insulin & acute exercise. Metab. Brain Dis. 2021, 36, 1289–1303.
- Aponte Becerra, L.; Galindo Mendez, B.; Khan, F.; Lioutas, V.; Novak, P.; Mantzoros, C.S.; Ngo, L.H.; Novak, V. Safety of Intranasal Insulin in Type 2 Diabetes on Systemic Insulin: A Double-Blinded Placebo-Controlled Sub-Study of Memaid Trial. Arch. Diabetes Obes. 2022, 4, 403–415.
- Schneider, E.; Spetter, M.S.; Martin, E.; Sapey, E.; Yip, K.P.; Manolopoulos, K.N.; Tahrani, A.A.; Thomas, J.M.; Lee, M.; Hallschmid, M.; et al. The effect of intranasal insulin on appetite and mood in women with and without obesity: An experimental medicine study. Int. J. Obes. 2022, 46, 1319–1327.
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454.
- Schechter, R.; Whitmire, J.; Wheet, G.S.; Beju, D.; Jackson, K.W.; Harlow, R.; Gavin, J.R., III. Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res. 1994, 636, 9–27.
- Gerozissis, K. Brain insulin: Regulation, mechanisms of action and functions. Cell. Mol. Neurobiol. 2003, 23, 1–25.
- Gerozissis, K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur. J. Pharmacol. 2008, 585, 38–49.
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161.
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521.
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487.
- Yu, Y.; Kastin, A.J.; Pan, W. Reciprocal interactions of insulin and insulin-like growth factor I in receptor-mediated transport across the blood-brain barrier. Endocrinology 2006, 147, 2611–2615.
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Effect of diabetes mellitus on the permeability of the blood-brain barrier to insulin. Peptides 1997, 18, 1577–1584.
- Banks, W.A.; Dohgu, S.; Lynch, J.L.; Fleegal-DeMotta, M.A.; Erickson, M.A.; Nakaoke, R.; Vo, T.Q. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology 2008, 149, 1514–1523.
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597.
- May, A.A.; Bedel, N.D.; Shen, L.; Woods, S.C.; Liu, M. Estrogen and insulin transport through the blood-brain barrier. Physiol. Behav. 2016, 163, 312–321.
- May, A.A.; Liu, M.; Woods, S.C.; Begg, D.P. CCK increases the transport of insulin into the brain. Physiol. Behav. 2016, 165, 392–397.
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399.
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314.
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930.
- Beddows, C.A.; Dodd, G.T. Insulin on the brain: The role of central insulin signalling in energy and glucose homeostasis. J. Neuroendocrinol. 2021, 33, e12947.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561.
- Villaseñor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Grüninger, F.; Loetscher, H.; Freskgård, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. J. Cereb. Blood Flow Metab. 2017, 37, 3683–3694.
- Mäe, M.A.; He, L.; Nordling, S.; Vazquez-Liebanas, E.; Nahar, K.; Jung, B.; Li, X.; Tan, B.C.; Chin Foo, J.; Cazenave-Gassiot, A.; et al. Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circ. Res. 2021, 128, e46–e62.
- McKinley, M.J.; Denton, D.A.; Ryan, P.J.; Yao, S.T.; Stefanidis, A.; Oldfield, B.J. From sensory circumventricular organs to cerebral cortex: Neural pathways controlling thirst and hunger. J. Neuroendocrinol. 2019, 31, e12689.
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962.
- Porniece Kumar, M.; Cremer, A.L.; Klemm, P.; Steuernagel, L.; Sundaram, S.; Jais, A.; Hausen, A.C.; Tao, J.; Secher, A.; Pedersen, T.Å.; et al. Insulin signalling in tanycytes gates hypothalamic insulin uptake and regulation of AgRP neuron activity. Nat. Metab. 2021, 3, 1662–1679.
- Langlet, F.; Levin, B.E.; Luquet, S.; Mazzone, M.; Messina, A.; Dunn-Meynell, A.A.; Balland, E.; Lacombe, A.; Mazur, D.; Carmeliet, P.; et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell. Metab. 2013, 17, 607–617.
- Prevot, V.; Langlet, F.; Dehouck, B. Flipping the tanycyte switch: How circulating signals gain direct access to the metabolic brain. Aging 2013, 5, 332–334.
- Morita-Takemura, S.; Wanaka, A. Blood-to-brain communication in the hypothalamus for energy intake regulation. Neurochem. Int. 2019, 28, 135–142.
- Dorn, A.; Bernstein, H.G.; Hahn, H.J.; Ziegler, M.; Rummelfanger, H. Insulin immunohistochemistry of rodent CNS: Apparent species differences but good correlation with radioimmunological data. Histochemistry 1981, 71, 609–616.
- Dorn, A.; Bernstein, H.G.; Rinne, A.; Hahn, H.J.; Ziegler, M. Insulin-like immunoreactivity in the human brain—A preliminary report. Histochemistry 1982, 74, 293–300.
- Baskin, D.G.; Brewitt, B.; Davidson, D.A.; Corp, E.; Paquette, T.; Figlewicz, D.P.; Lewellen, T.K.; Graham, M.K.; Woods, S.G.; Dorsa, D.M. Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes 1986, 35, 246–249.
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic localization of insulin receptors in rat brain: Prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138.
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570.
- Unger, J.; McNeill, T.H.; Moxley, R.T., 3rd; White, M.; Moss, A.; Livingston, J.N. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 1989, 31, 143–157.
- Marks, J.L.; Porte, D., Jr.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236.
- Iozzo, P.; Osman, S.; Glaser, M.; Knickmeier, M.; Ferrannini, E.; Pike, V.W.; Camici, P.G.; Law, M.P. In vivo imaging of insulin receptors by PET: Preclinical evaluation of iodine-125 and iodine-124 labelled human insulin. Nucl. Med. Biol. 2002, 29, 73–82.
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81.
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin action on astrocytes: From energy homeostasis to behaviour. J. Neuroendocrinol. 2021, 33, e12953.
- Moller, D.E.; Yokota, A.; Caro, J.F.; Flier, J.S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 1989, 3, 1263–1269.
- Seino, S.; Bell, G.I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 1989, 159, 312–316.
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413.
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118.
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431.
- Escribano, O.; Beneit, N.; Rubio-Longás, C.; López-Pastor, A.R.; Gómez-Hernández, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206.
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221.
- Drakas, R.; Tu, X.; Baserga, R. Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 9272–9276.
- De Meyts, P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 2008, 33, 376–384.
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623.
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304.
- Razzini, G.; Ingrosso, A.; Brancaccio, A.; Sciacchitano, S.; Esposito, D.L.; Falasca, M. Different subcellular localization and phosphoinositides binding of insulin receptor substrate protein pleckstrin homology domains. Mol. Endocrinol. 2000, 14, 823–836.
- Sadagurski, M.; Dong, X.C.; Myers, M.G., Jr.; White, M.F. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2013, 3, 55–63.
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243.
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582.
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341.
- Levenga, J.; Wong, H.; Milstead, R.A.; Keller, B.N.; LaPlante, L.E.; Hoeffer, C.A. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. eLife 2017, 6, e30640.
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405.
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189.
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911.
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776.
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131.
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192.
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimers Dis. 2011, 2011, 189728.
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848.
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105.
- Kim, K.W.; Donato, J., Jr.; Berglund, E.D.; Choi, Y.H.; Kohno, D.; Elias, C.F.; Depinho, R.A.; Elmquist, J.K. FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Invest. 2012, 122, 2578–2589.
- Ren, H.; Plum-Morschel, L.; Gutierrez-Juarez, R.; Lu, T.Y.; Kim-Muller, J.Y.; Heinrich, G.; Wardlaw, S.L.; Silver, R.; Accili, D. Blunted refeeding response and increased locomotor activity in mice lacking FoxO1 in synapsin-Cre-expressing neurons. Diabetes 2013, 62, 3373–3383.
- Du, S.; Zheng, H. Role of FoxO transcription factors in aging and age-related metabolic and neurodegenerative diseases. Cell Biosci. 2021, 11, 188.
- Oli, V.; Gupta, R.; Kumar, P. FOXO and related transcription factors binding elements in the regulation of neurodegenerative disorders. J. Chem. Neuroanat. 2021, 116, 102012.
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171.
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology 2006, 21, 362–369.
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796.
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234.
- Bakke, J.; Haj, F.G. Protein-tyrosine phosphatase 1B substrates and metabolic regulation. Semin. Cell Dev. Biol. 2015, 37, 58–65.
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674.
- Dodd, G.T.; Xirouchaki, C.E.; Eramo, M.; Mitchell, C.A.; Andrews, Z.B.; Henry, B.A.; Cowley, M.A.; Tiganis, T. Intranasal Targeting of Hypothalamic PTP1B and TCPTP Reinstates Leptin and Insulin Sensitivity and Promotes Weight Loss in Obesity. Cell Rep. 2019, 28, 2905–2922.e5.
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537.
- Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317.
- Nogueiras, R.; Sabio, G. Brain JNK and metabolic disease. Diabetologia 2021, 64, 265–274.
- Busquets, O.; Espinosa-Jiménez, T.; Ettcheto, M.; Olloquequi, J.; Bulló, M.; Carro, E.; Cantero, J.L.; Casadesús, G.; Folch, J.; Verdaguer, E.; et al. JNK1 and JNK3: Divergent functions in hippocampal metabolic-cognitive function. Mol. Med. 2022, 28, 48.
- Accardi, G.; Virruso, C.; Balistreri, C.R.; Emanuele, F.; Licastro, F.; Monastero, R.; Porcellini, E.; Vasto, S.; Verga, S.; Caruso, C.; et al. SHIP2: A "new" insulin pathway target for aging research. Rejuvenation Res. 2014, 17, 221–225.
- Sekar, S.; Taghibiglou, C. Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci. Lett. 2018, 666, 139–143.
- Zhao, J.; Yin, L.; Jiang, L.; Hou, L.; He, L.; Zhang, C. PTEN nuclear translocation enhances neuronal injury after hypoxia-ischemia via modulation of the nuclear factor-κB signaling pathway. Aging 2021, 13, 16165–16177.
- Javadpour, P.; Dargahi, L.; Ahmadiani, A.; Ghasemi, R. To be or not to be: PP2A as a dual player in CNS functions, its role in neurodegeneration, and its interaction with brain insulin signaling. Cell. Mol. Life Sci. 2019, 76, 2277–2297.
- Frey, W.H., II. Neurologic Agents for Nasal Administration to the Brain. PCT International Patent WO 91/07947, 13 June 1991.
- Frey, W.H., II. Method of Administering Neurologic Agents to the Brain. U.S. Patent 5,624,898, 29 April 1997.
- Frey, W.H., II. Method for Administering Insulin to the Brain. U.S. Patent 6,313,093 B1, 6 November 2001.
- Nedelcovych, M.T.; Gadiano, A.J.; Wu, Y.; Manning, A.A.; Thomas, A.G.; Khuder, S.S.; Yoo, S.W.; Xu, J.; McArthur, J.C.; Haughey, N.J.; et al. Pharmacokinetics of Intranasal versus Subcutaneous Insulin in the Mouse. ACS Chem. Neurosci. 2018, 9, 809–816.
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309.
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628.
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H., II. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496.
- Salameh, T.S.; Bullock, K.M.; Hujoel, I.A.; Niehoff, M.L.; Wolden-Hanson, T.; Kim, J.; Morley, J.E.; Farr, S.A.; Banks, W.A. Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J. Alzheimers Dis. 2015, 47, 715–728.
- Chudoba, C.; Kleinridders, A. 271-LB: Intranasal Insulin Treatment Causes Sex-Specific Differences in Metabolism and Behavior in Mice. Diabetes 2019, 68 (Suppl. 1), 271-LB.
- Jauch-Chara, K.; Friedrich, A.; Rezmer, M.; Melchert, U.H.; Scholand-Engler, H.G.; Hallschmid, M.; Oltmanns, K.M. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes 2012, 61, 2261–2268.
- Leary, A.C.; Stote, R.M.; Breedt, H.J.; O’Brien, J.; Buckley, B. Pharmacokinetics and pharmacodynamics of intranasal insulin administered to healthy subjects in escalating doses. Diabetes Technol. Ther. 2005, 7, 124–130.
- Leker, R.R.; Shohami, E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: Neuroprotective opportunities. Brain Res. Brain Res. Rev. 2002, 39, 55–73.
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G.; et al. Stroke Thrombolysis Trialists’ Collaborative Group. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: A meta-analysis of individual patient data from randomised trials. Lancet 2014, 384, 1929–1935.
- Zhou, Z.B.; Meng, L.; Gelb, A.W.; Lee, R.; Huang, W.Q. Cerebral ischemia during surgery: An overview. J. Biomed. Res. 2016, 30, 83–87.
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215.
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J. Neuroinflamm. 2010, 7, 14.
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966.
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107.
- Dharmasaroja, P.A. Fluid Intake Related to Brain Edema in Acute Middle Cerebral Artery Infarction. Transl. Stroke Res. 2016, 7, 49–53.
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients with Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418.
- Greenberg, S.M.; Ziai, W.C.; Cordonnier, C.; Dowlatshahi, D.; Francis, B.; Goldstein, J.N.; Hemphill, J.C., 3rd; Johnson, R.; Keigher, K.M.; Mack, W.J.; et al. American Heart Association/American Stroke Association. 2022 Guideline for the Management of Patients with Spontaneous Intracerebral Hemorrhage: A Guideline from the American Heart Association/American Stroke Association. Stroke 2022, 53, e282–e361.
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432.
- Zhang, Z.; Yan, J.; Shi, H. Hyperglycemia as a Risk Factor of Ischemic Stroke. J. Drug Metab. Toxicol. 2013, 4, 153.
- Palaiodimou, L.; Lioutas, V.A.; Lambadiari, V.; Paraskevas, G.P.; Voumvourakis, K.; Tsivgoulis, G. Glycemia management in acute ischemic stroke: Current concepts and novel therapeutic targets. Postgrad. Med. 2019, 131, 423–437.
- Middleton, S.; McElduff, P.; Ward, J.; Grimshaw, J.M.; Dale, S.; D’Este, C.; Drury, P.; Griffiths, R.; Cheung, N.W.; Quinn, C.; et al. QASC Trialists Group. Implementation of evidence-based treatment protocols to manage fever, hyperglycaemia, and swallowing dysfunction in acute stroke (QASC): A cluster randomised controlled trial. Lancet 2011, 378, 1699–1706.
- Passero, S.; Ciacci, G.; Ulivelli, M. The influence of diabetes and hyperglycemia on clinical course after intracerebral hemorrhage. Neurology 2003, 61, 1351–1356.
- Kim, Y.; Han, M.H.; Kim, C.H.; Kim, J.M.; Cheong, J.H.; Ryu, J.I. Increased Short-Term Mortality in Patients with Spontaneous Intracerebral Hemorrhage and its Association with Admission Glucose Levels and Leukocytosis. World Neurosurg. 2017, 98, 503–511.
- Almdal, T.; Scharling, H.; Jensen, J.S.; Vestergaard, H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: A population-based study of 13,000 men and women with 20 years of follow-up. Arch. Intern. Med. 2004, 164, 1422–1426.
- Banerjee, C.; Moon, Y.P.; Paik, M.C.; Rundek, T.; Mora-McLaughlin, C.; Vieira, J.R.; Sacco, R.L.; Elkind, M.S. Duration of diabetes and risk of ischemic stroke: The Northern Manhattan Study. Stroke 2012, 43, 1212–1217.
- Mitsios, J.P.; Ekinci, E.I.; Mitsios, G.P.; Churilov, L.; Thijs, V. Relationship Between Glycated Hemoglobin and Stroke Risk: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e007858.
- Pan, Y.; Chen, W.; Wang, Y. Prediabetes and Outcome of Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Meta-analysis. J. Stroke Cerebrovasc. Dis. 2019, 28, 683–692.
- Kernan, W.N.; Inzucchi, S.E.; Viscoli, C.M.; Brass, L.M.; Bravata, D.M.; Shulman, G.I.; McVeety, J.C.; Horwitz, R.I. Impaired insulin sensitivity among nondiabetic patients with a recent TIA or ischemic stroke. Neurology 2003, 60, 1447–1451.
- Hishinuma, A.; Majima, M.; Kurabayashi, H. Insulin resistance in patients with stroke is related to visceral fat obesity and adipocytokines. J. Stroke Cerebrovasc. Dis. 2008, 17, 175–180.
- Voll, C.L.; Auer, R.N. The effect of postischemic blood glucose levels on ischemic brain damage in the rat. Ann. Neurol. 1988, 24, 638–646.
- Voll, C.L.; Auer, R.N. Insulin attenuates ischemic brain damage independent of its hypoglycemic effect. J. Cereb. Blood Flow Metab. 1991, 11, 1006–1014.
- Voll, C.L.; Whishaw, I.Q.; Auer, R.N. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke 1989, 20, 646–651.
- Hamilton, M.G.; Tranmer, B.I.; Auer, R.N. Insulin reduction of cerebral infarction due to transient focal ischemia. J. Neurosurg. 1995, 82, 262–268.
- Guyot, L.L.; Diaz, F.G.; O’Regan, M.H.; Ren, J.; Phillis, J.W. The effect of intravenous insulin on accumulation of excitotoxic and other amino acids in the ischemic rat cerebral cortex. Neurosci. Lett. 2000, 288, 61–65.
- Fukuoka, S.; Yeh, H.; Mandybur, T.I.; Tew, J.M., Jr. Effect of insulin on acute experimental cerebral ischemia in gerbils. Stroke 1989, 20, 396–399.
- Meden, P.; Andersen, M.; Overgaard, K.; Rasmussen, R.S.; Boysen, G. The effects of early insulin treatment combined with thrombolysis in rat embolic stroke. Neurol. Res. 2002, 24, 399–404.
- Huang, S.S.; Lu, Y.J.; Huang, J.P.; Wu, Y.T.; Day, Y.J.; Hung, L.M. The essential role of endothelial nitric oxide synthase activation in insulin-mediated neuroprotection against ischemic stroke in diabetes. J. Vasc. Surg. 2014, 59, 483–491.
- Izumi, Y.; Pinard, E.; Roussel, S.; Seylaz, J. Insulin protects brain tissue against focal ischemia in rats. Neurosci. Lett. 1992, 144, 121–123.
- Fanne, R.A.; Nassar, T.; Heyman, S.N.; Hijazi, N.; Higazi, A.A. Insulin and glucagon share the same mechanism of neuroprotection in diabetic rats: Role of glutamate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R668–R673.
- Guan, J.; Williams, C.; Gunning, M.; Mallard, C.; Gluckman, P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J. Cereb. Blood Flow Metab. 1993, 13, 609–616.
- Zhu, C.Z.; Auer, R.N. Intraventricular administration of insulin and IGF-1 in transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 237–242.
- Russo, V.; Candeloro, P.; Malara, N.; Perozziello, G.; Iannone, M.; Scicchitano, M.; Mollace, R.; Musolino, V.; Gliozzi, M.; Carresi, C.; et al. Key Role of Cytochrome C for Apoptosis Detection Using Raman Microimaging in an Animal Model of Brain Ischemia with Insulin Treatment. Appl. Spectrosc. 2019, 73, 1208–1217.
- Sanderson, T.H.; Kumar, R.; Sullivan, J.M.; Krause, G.S. Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-XL binding. J. Neurochem. 2008, 106, 1248–1258.
- Sanderson, T.H.; Kumar, R.; Murariu-Dobrin, A.C.; Page, A.B.; Krause, G.S.; Sullivan, J.M. Insulin activates the PI3K-Akt survival pathway in vulnerable neurons following global brain ischemia. Neurol. Res. 2009, 31, 947–958.
- Duarte, A.I.; Santos, P.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 2008, 1783, 994–1002.
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017.
- McKay, M.K.; Hester, R.L. Role of nitric oxide, adenosine, and ATP-sensitive potassium channels in insulin-induced vasodilation. Hypertension 1996, 28, 202–208.
- Hung, L.M.; Huang, J.P.; Liao, J.M.; Yang, M.H.; Li, D.E.; Day, Y.J.; Huang, S.S. Insulin renders diabetic rats resistant to acute ischemic stroke by arresting nitric oxide reaction with superoxide to form peroxynitrite. J. Biomed. Sci. 2014, 21, 92.
- Hughes, T.M.; Craft, S. The role of insulin in the vascular contributions to age-related dementia. Biochim. Biophys. Acta 2016, 1862, 983–991.
- Muniyappa, R.; Yavuz, S. Metabolic actions of angiotensin II and insulin: A microvascular endothelial balancing act. Mol. Cell. Endocrinol. 2013, 378, 59–69.
- Akintola, A.A.; van Opstal, A.M.; Westendorp, R.G.; Postmus, I.; van der Grond, J.; van Heemst, D. Effect of intranasally administered insulin on cerebral blood flow and perfusion; a randomized experiment in young and older adults. Aging 2017, 9, 790–802.
- Lioutas, V.A.; Alfaro-Martinez, F.; Bedoya, F.; Chung, C.C.; Pimentel, D.A.; Novak, V. Intranasal Insulin and Insulin-Like Growth Factor 1 as Neuroprotectants in Acute Ischemic Stroke. Transl. Stroke Res. 2015, 6, 264–275.
- Zorina, I.I.; Zakharova, I.O.; Bayunova, L.V.; Avrova, N.F. Insulin Administration Prevents Accumulation of Conjugated Dienes and Trienes and Inactivation of Na+, K+-ATPase in the Rat Cerebral Cortex during Two-Vessel Forebrain Ischemia and Reperfusion. J. Evol. Biochem. Physiol. 2018, 54, 246–249.
- Zorina, I.I.; Galkina, O.V.; Bayunova, L.V.; Zakharova, I.O. Effect of Insulin on Lipid Peroxidation and Glutathione Levels in a Two-Vessel Occlusion Model of Rat Forebrain Ischemia Followed by Reperfusion. J. Evol. Biochem. Physiol. 2019, 55, 333–335.
- Zakharova, I.O.; Bayunova, L.V.; Zorina, I.I.; Sokolova, T.V.; Shpakov, A.O.; Avrova., N.F. Insulin and α-Tocopherol Enhance the Protective Effect of Each Other on Brain Cortical Neurons under Oxidative Stress Conditions and in Rat Two-Vessel Forebrain Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 11768.
- Zakharova, I.O.; Bayunova, L.V.; Zorina, I.I.; Shpakov, A.O.; Avrova, N.F. Insulin and Brain Gangliosides Prevent Metabolic Disorders Caused by Activation of Free Radical Reactions after Two-Vessel Ischemia–Reperfusion Injury to the Rat Forebrain. J. Evol. Biochem. Physiol. 2022, 58, 279–291.
- Xu, L.B.; Huang, H.D.; Zhao, M.; Zhu, G.C.; Xu, Z. Intranasal Insulin Treatment Attenuates Metabolic Distress and Early Brain Injury After Subarachnoid Hemorrhage in Mice. Neurocrit. Care 2021, 34, 154–166.
- Zhu, Y.; Huang, Y.; Yang, J.; Tu, R.; Zhang, X.; He, W.W.; Hou, C.Y.; Wang, X.M.; Yu, J.M.; Jiang, G.H. Intranasal insulin ameliorates neurological impairment after intracerebral hemorrhage in mice. Neural Regen. Res. 2022, 17, 210–216.
- Liu, X.F.; Fawcett, J.R.; Thorne, R.G.; DeFor, T.A.; Frey, W.H., II. Intranasal administration of insulin-like growth factor-I bypasses the blood-brain barrier and protects against focal cerebral ischemic damage. J. Neurol. Sci. 2001, 187, 91–97.
- Liu, X.F.; Fawcett, J.R.; Thorne, R.G.; Frey, W.H., II. Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci. Lett. 2001, 308, 91–94.
- Liu, X.F.; Fawcett, J.R.; Hanson, L.R.; Frey, W.H., II. The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin-like growth factor-I in rats. J. Stroke Cerebrovasc. Dis. 2004, 13, 16–23.
- Fletcher, L.; Kohli, S.; Sprague, S.M.; Scranton, R.A.; Lipton, S.A.; Parra, A.; Jimenez, D.F.; Digicaylioglu, M. Intranasal delivery of erythropoietin plus insulin-like growth factor-I for acute neuroprotection in stroke. Laboratory investigation. J. Neurosurg. 2009, 111, 164–170.
- Cai, Z.; Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G. Intranasal administration of insulin-like growth factor-1 protects against lipopolysaccharide-induced injury in the developing rat brain. Neuroscience 2011, 194, 195–207.
- Lin, S.; Rhodes, P.G.; Cai, Z. Whole body hypothermia broadens the therapeutic window of intranasally administered IGF-1 in a neonatal rat model of cerebral hypoxia-ischemia. Brain Res. 2011, 1385, 246–256.
- Shen, H.; Gu, X.; Wei, Z.Z.; Wu, A.; Liu, X.; Wei, L. Combinatorial intranasal delivery of bone marrow mesenchymal stem cells and insulin-like growth factor-1 improves neurovascularization and functional outcomes following focal cerebral ischemia in mice. Exp. Neurol. 2021, 337, 113542.
- Iravanpour, F.; Dargahi, L.; Rezaei, M.; Haghani, M.; Heidari, R.; Valian, N.; Ahmadiani, A. Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson’s disease. CNS Neurosci. Ther. 2021, 27, 308–319.
- Kim, B.H.; Kelschenbach, J.; Borjabad, A.; Hadas, E.; He, H.; Potash, M.J.; Nedelcovych, M.T.; Rais, R.; Haughey, N.J.; McArthur, J.C.; et al. Intranasal insulin therapy reverses hippocampal dendritic injury and cognitive impairment in a model of HIV-associated neurocognitive disorders in EcoHIV-infected mice. AIDS 2019, 33, 973–984.
- Rassovsky, Y.; Levi, Y.; Agranov, E.; Sela-Kaufman, M.; Sverdlik, A.; Vakil, E. Predicting long-term outcome following traumatic brain injury (TBI). J. Clin. Exp. Neuropsychol. 2015, 37, 354–366.
- Røe, C.; Sveen, U.; Alvsåker, K.; Bautz-Holter, E. Post-concussion symptoms after mild traumatic brain injury: Influence of demographic factors and injury severity in a 1-year cohort study. Disabil. Rehabil. 2009, 31, 1235–1243.
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75, S24–S33.
- Gross, H.; Kling, A.; Henry, G.; Herndon, C.; Lavretsky, H. Local cerebral glucose metabolism in patients with long-term behavioral and cognitive deficits following mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 1996, 8, 324–334.
- Toth, A. Magnetic Resonance Imaging Application in the Area of Mild and Acute Traumatic Brain Injury: Implications for Diagnostic Markers? In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2015; Chapter 24; ISBN 978-1-4665-6598-2.
- Llompart-Pou, J.A.; Raurich, J.M.; Pérez-Bárcena, J.; Barceló, A.; Ibáñez, J.; Ayestarán, J.I. Acute Hypothalamic-pituitary-adrenal response in traumatic brain injury with and without extracerebral trauma. Neurocrit. Care 2008, 9, 230–236.
- Shaughness, M.; Acs, D.; Brabazon, F.; Hockenbury, N.; Byrnes, K.R. Role of Insulin in Neurotrauma and Neurodegeneration: A Review. Front. Neurosci. 2020, 14, 547175.
- Arora, P.; Singh, K.; Kumari, M.; Trivedi, R. Temporal profile of serum metabolites and inflammation following closed head injury in rats is associated with HPA axis hyperactivity. Metabolomics 2022, 18, 28.
- Karelina, K.; Sarac, B.; Freeman, L.M.; Gaier, K.R.; Weil, Z.M. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 2016, 43, 1034–1043.
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228.
- Selwyn, R.; Hockenbury, N.; Jaiswal, S.; Mathur, S.; Armstrong, R.C.; Byrnes, K.R. Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J. Neurotrauma 2013, 30, 1943–1953.
- Yoshino, A.; Hovda, D.A.; Kawamata, T.; Katayama, Y.; Becker, D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991, 561, 106–119.
- Brabazon, F.; Wilson, C.M.; Shukla, D.K.; Mathur, S.; Jaiswal, S.; Bermudez, S.; Byrnes, K.R.; Selwyn, R. FDG-PET Combined with MRI Elucidates the Pathophysiology of Traumatic Brain Injury in Rats. J. Neurotrauma 2017, 34, 1074–1085.
- Yasmin, A.; Jokivarsi, K.; Poutiainen, P.; Pitkänen, A.; Gröhn, O.; Immonen, R. Chronic hypometabolism in striatum and hippocampal network after traumatic brain injury and their relation with memory impairment—-FDG-PET and MRI 4 months after fluid percussion injury in rat. Brain Res. 2022, 1788, 147934.
- Brabazon, F.; Wilson, C.M.; Jaiswal, S.; Reed, J.; Frey, W.H., 2nd; Byrnes, K.R. Intranasal insulin treatment of an experimental model of moderate traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 3203–3218.
- Zhang, J.; Zhang, Y.; Zou, J.; Cao, F. A meta-analysis of cohort studies: Traumatic brain injury and risk of Alzheimer’s Disease. PLoS ONE 2021, 16, e0253206.
- Colca, J.R.; Finck, B.N. Metabolic Mechanisms Connecting Alzheimer’s and Parkinson’s Diseases: Potential Avenues for Novel Therapeutic Approaches. Front. Mol. Biosci. 2022, 9, 929328.
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cereb. Blood Flow Metab. 2004, 24, 133–150.
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Prim. 2018, 4, 18003.
- Nijssen, K.M.R.; Mensink, R.P.; Joris, P.J. Effects of Intranasal Insulin Administration on Cerebral Blood Flow and Cognitive Performance in Adults: A Systematic Review of Randomized, Placebo-Controlled Intervention Studies. Neuroendocrinology 2022, 24, 1–13.
- Blázquez, E.; Hurtado-Carneiro, V.; LeBaut-Ayuso, Y.; Velázquez, E.; García-García, L.; Gómez-Oliver, F.; Ruiz-Albusac, J.M.; Ávila, J.; Pozo, M.Á. Significance of Brain Glucose Hypometabolism, Altered Insulin Signal Transduction, and Insulin Resistance in Several Neurological Diseases. Front. Endocrinol. 2022, 13, 873301.
- Lee, T.H.; Yau, S.Y. From Obesity to Hippocampal Neurodegeneration: Pathogenesis and Non-Pharmacological Interventions. Int. J. Mol. Sci. 2020, 22, 201.
- Sharma, S. High fat diet and its effects on cognitive health: Alterations of neuronal and vascular components of brain. Physiol. Behav. 2021, 240, 113528.
- Al Haj Ahmad, R.M.; Ababneh, N.A.; Al-Domi, H.A. Brain insulin resistance as a mechanistic mediator links peripheral metabolic disorders with declining cognition. Diabetes Metab. Syndr. 2022, 16, 102468.
- Dutta, B.J.; Singh, S.; Seksaria, S.; Das Gupta, G.; Singh, A. Inside the diabetic brain: Insulin resistance and molecular mechanism associated with cognitive impairment and its possible therapeutic strategies. Pharmacol. Res. 2022, 182, 106358.
- Zhang, Q.; Jin, K.; Chen, B.; Liu, R.; Cheng, S.; Zhang, Y.; Lu, J. Overnutrition Induced Cognitive Impairment: Insulin Resistance, Gut-Brain Axis, and Neuroinflammation. Front. Neurosci. 2022, 16, 884579.
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181.
- Schulingkamp, R.J.; Pagano, T.C.; Hung, D.; Raffa, R.B. Insulin receptors and insulin action in the brain: Review and clinical implications. Neurosci. Biobehav. Rev. 2000, 24, 855–872.
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711.
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Häring, H.U.; Fritsche, A.; Preissl, H. Selective insulin resistance in homeostatic and cognitive control brain areas in overweight and obese adults. Diabetes Care 2015, 38, 1044–1050.
- Brands, A.M.; Kessels, R.P.; de Haan, E.H.; Kappelle, L.J.; Biessels, G.J. Cerebral dysfunction in type 1 diabetes: Effects of insulin, vascular risk factors and blood-glucose levels. Eur. J. Pharmacol. 2004, 490, 159–168.
- Wicha, P.; Das, S.; Mahakkanukrauh, P. Blood-brain barrier dysfunction in ischemic stroke and diabetes: The underlying link, mechanisms and future possible therapeutic targets. Anat. Cell Biol. 2021, 54, 165–177.
- Romanova, I.V.; Derkach, K.V.; Mikhrina, A.L.; Sukhov, I.B.; Mikhailova, E.V.; Shpakov, A.O. The Leptin, Dopamine and Serotonin Receptors in Hypothalamic POMC-Neurons of Normal and Obese Rodents. Neurochem. Res. 2018, 43, 821–837.
- Derkach, K.; Zakharova, I.; Zorina, I.; Bakhtyukov, A.; Romanova, I.; Bayunova, L.; Shpakov, A. The evidence of metabolic-improving effect of metformin in Ay/a mice with genetically-induced melanocortin obesity and the contribution of hypothalamic mechanisms to this effect. PLoS ONE 2019, 14, e0213779.
- Schuh, A.F.; Rieder, C.M.; Rizzi, L.; Chaves, M.; Roriz-Cruz, M. Mechanisms of brain aging regulation by insulin: Implications for neurodegeneration in late-onset Alzheimer’s disease. ISRN Neurol. 2011, 2011, 306905.
- Matioli, M.N.P.S.; Nitrini, R. Mechanisms linking brain insulin resistance to Alzheimer’s disease. Dement. Neuropsychol. 2015, 9, 96–102.
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357.
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvourakis, K.I.; et al. Type 2 Diabetes Mellitus as a Risk Factor for Alzheimer’s Disease: Review and Meta-Analysis. Biomedicines 2022, 10, 778.
- Santiago, J.C.P.; Hallschmid, M. Outcomes and clinical implications of intranasal insulin administration to the central nervous system. Exp. Neurol. 2019, 317, 180–190.
- Riederer, P.; Korczyn, A.D.; Ali, S.S.; Bajenaru, O.; Choi, M.S.; Chopp, M.; Dermanovic-Dobrota, V.; Grünblatt, E.; Jellinger, K.A.; Kamal, M.A.; et al. The diabetic brain and cognition. J. Neural Transm. 2017, 124, 1431–1454.
- Li, T.; Cao, H.X.; Ke, D. Type 2 Diabetes Mellitus Easily Develops into Alzheimer’s Disease via Hyperglycemia and Insulin Resistance. Curr. Med. Sci. 2021, 41, 1165–1171.
- Novak, V.; Milberg, W.; Hao, Y.; Munshi, M.; Novak, P.; Galica, A.; Manor, B.; Roberson, P.; Craft, S.; Abduljalil, A. Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care 2014, 37, 751–759.
- Zhang, H.; Hao, Y.; Manor, B.; Novak, P.; Milberg, W.; Zhang, J.; Fang, J.; Novak, V. Intranasal insulin enhanced resting-state functional connectivity of hippocampal regions in type 2 diabetes. Diabetes 2015, 64, 1025–1034.
- Wang, Y.; Sun, L.; He, G.; Gang, X.; Zhao, X.; Wang, G.; Ning, G. Cerebral perfusion alterations in type 2 diabetes mellitus—A systematic review. Front. Neuroendocrinol. 2021, 62, 100916.
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166.
- Caplan, G.A.; Lan, Z.; Newton, L.; Kvelde, T.; McVeigh, C.; Hill, M.A. Transcranial Doppler to measure cerebral blood flow in delirium superimposed on dementia. A cohort study. J. Am. Med. Dir. Assoc. 2014, 15, 355–360.
- Maldonado, J.R. Delirium pathophysiology: An updated hypothesis of the etiology of acute brain failure. Int. J. Geriatr. Psychiatry 2018, 33, 1428–1457.
- Caplan, G.A.; Kvelde, T.; Lai, C.; Yap, S.L.; Lin, C.; Hill, M.A. Cerebrospinal fluid in long-lasting delirium compared with Alzheimer’s dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1130–1136.
- Haggstrom, L.R.; Nelson, J.A.; Wegner, E.A.; Caplan, G.A. 2-18F-fluoro-2-deoxyglucose positron emission tomography in delirium. J. Cereb. Blood Flow Metab. 2017, 37, 3556–3567.
- Ma, H.; Liao, Y.; Mo, Y.; Li, Z.-R.; Liao, Q.; Wang, Y.-C.; Duan, K.-M.; Chen, M.-H.; Ouyang, W. Decreased cerebral glucose metabolism in elderly patients with postoperative delirium: A case-control study. J. Anesth. Perioper. Med. 2017, 4, 162–168.
- Evered, L.; Silbert, B.; Knopman, D.S.; Scott, D.A.; DeKosky, S.T.; Rasmussen, L.S.; Oh, E.S.; Crosby, G.; Berger, M.; Eckenhoff, R.G.; et al. Recommendations for the nomenclature of cognitive change associated with anaesthesia and surgery-2018. Br. J. Anaesth. 2018, 121, 1005–1012.
- Duning, T.; Ilting-Reuke, K.; Beckhuis, M.; Oswald, D. Postoperative delirium—Treatment and prevention. Curr. Opin. Anaesthesiol. 2021, 34, 27–32.
- Fu, D.; Tan, X.; Zhang, M.; Chen, L.; Yang, J. Association between frailty and postoperative delirium: A meta-analysis of cohort study. Aging Clin. Exp. Res. 2022, 34, 25–37.
- Needham, M.J.; Webb, C.E.; Bryden, D.C. Postoperative cognitive dysfunction and dementia: What we need to know and do. Br. J. Anaesth. 2017, 119, i115–i125.
- Shoair, O.A.; Grasso Ii, M.P.; Lahaye, L.A.; Daniel, R.; Biddle, C.J.; Slattum, P.W. Incidence and risk factors for postoperative cognitive dysfunction in older adults undergoing major noncardiac surgery: A prospective study. J. Anaesthesiol. Clin. Pharmacol. 2015, 31, 30–36.
- Borozdina, A.; Qeva, E.; Cinicola, M.; Bilotta, F. Perioperative cognitive evaluation. Curr. Opin. Anaesthesiol. 2018, 31, 756–761.
- Hermanides, J.; Qeva, E.; Preckel, B.; Bilotta, F. Perioperative hyperglycemia and neurocognitive outcome after surgery: A systematic review. Minerva Anestesiol. 2018, 84, 1178–1188.
- Badenes, R.; Qeva, E.; Giordano, G.; Romero-García, N.; Bilotta, F. Intranasal Insulin Administration to Prevent Delayed Neurocognitive Recovery and Postoperative Neurocognitive Disorder: A Narrative Review. Int. J. Environ. Res. Public Health 2021, 18, 2681.
- Neirijnck, Y.; Papaioannou, M.D.; Nef, S. The Insulin/IGF System in Mammalian Sexual Development and Reproduction. Int. J. Mol. Sci. 2019, 20, 4440.
- Oghbaei, H.; Fattahi, A.; Hamidian, G.; Sadigh-Eteghad, S.; Ziaee, M.; Mahmoudi, J. A closer look at the role of insulin for the regulation of male reproductive function. Gen. Comp. Endocrinol. 2021, 300, 113643.