 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Angeliki Kotsiafti | -- | 3439 | 2023-04-10 19:44:37 | | | |
| 2 | Peter Tang | -1 word(s) | 3438 | 2023-04-11 03:10:42 | | |
Video Upload Options
Therapy-related acute myeloid leukemia (t-AML) is one of the most serious long-term complications of cancer chemotherapy. Various cytotoxic agents and exposure to ionizing radiation can lead to the development of t-AML, which is usually associated with adverse genetic changes and a poor prognosis.
1. Introduction
2. Etiology of t-AML
|
Drug Class |
Mechanism of Action |
|---|---|
|
Alkylating Agents Cyclophosphamide, * cisplatin, carboplatin, melphalan, busulphan, chlorambucil, lomustin, carmustine, dacarbazine, procarbazine, thiotepa, mitomycin C |
Creation of bonds in one or both DNA strands, through alkylation |
|
Topoisomerase II Inhibitors Etoposide, teniposide, doxorubicin, idarubicin, daunorubicin, mitoxantrone, * actinomycin D, amsacrine |
“Topoisomerase II poisons” convert topoisomerase II into a DNA-damaging enzyme |
|
Antimetabolites Fludarabine, cladribine, * pentostatin, thiopurines (6-mercaptopurine, * 6-thioguanine, azathioprine *), mycophenolate mofetil * |
They act as mimics of other molecules, and in this way, they interfere with DNA and RNA synthesis |
|
Antitubulin Agents Vinblastine, vindesine, vincristine, docetaxel, paclitaxel |
Antimitotic agents that bind tubulin dimers and disrupt the formation of mitotic spindle |
|
Poly (ADP-Ribose) Polymerase (PARP) Inhibitors Olaparib, talazoparib, niraparib, rucaparib |
Inhibitors of the PARP family of enzymes inhibit homologous recombination repair (PARP enzymes, activated by DNA damage, repair the single-helix DNA breaks by forming branched PAR chains that serve as a docking platform for DNA repair enzymes) |
Abbreviations: t-AML, therapy-related acute myeloid leukemia; PARP, poly (ADP-ribose) polymerase. * Widely used as immunosuppressive agents in the treatment of autoimmune and immune-mediated inflammatory disorders.
2.1. Types of t-AML
3. Pathophysiology of t-AML
3.1. Genetic Predisposition for t-AML
3.2. Current Model for the Molecular Pathogenesis of t-AML
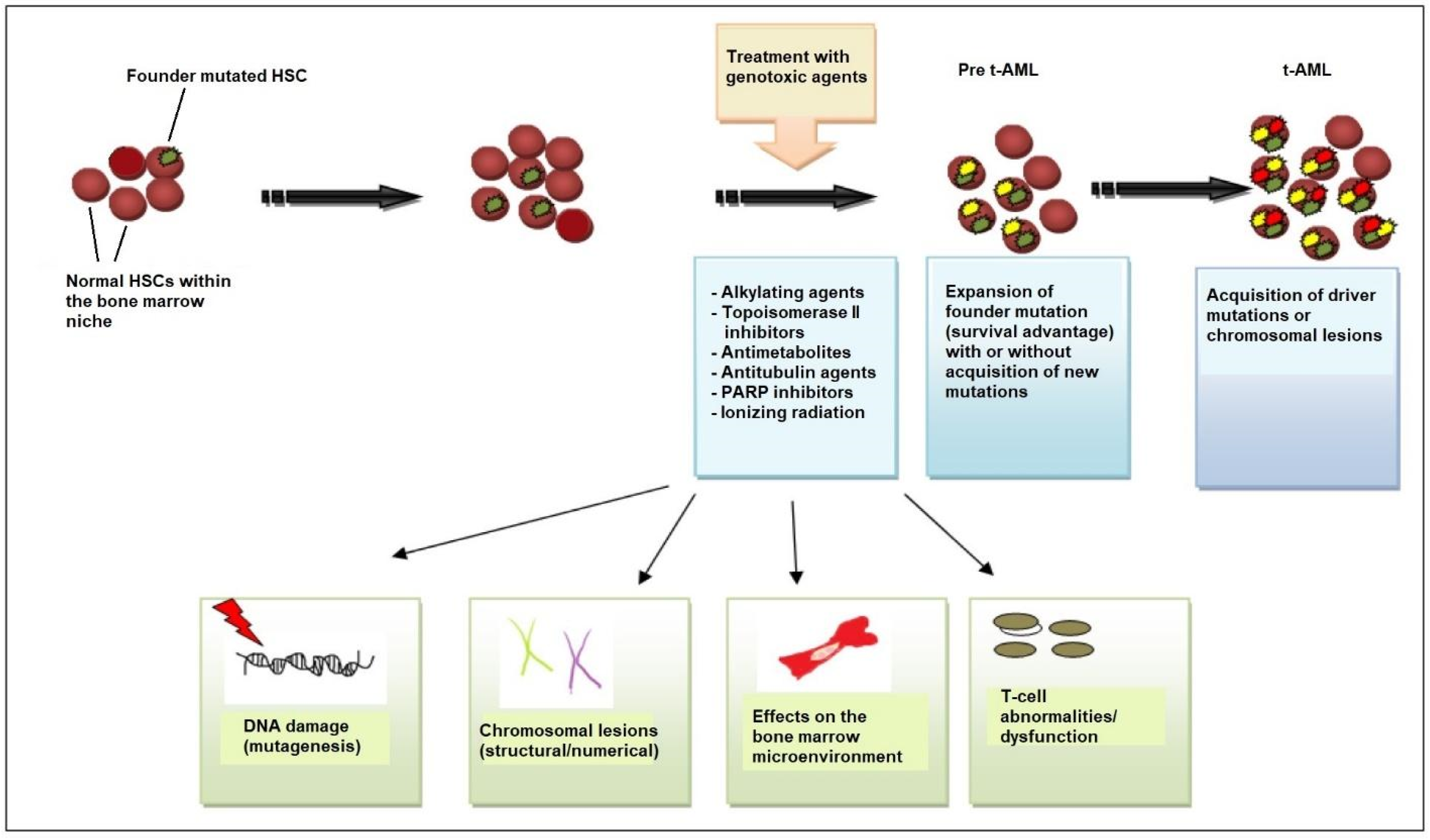
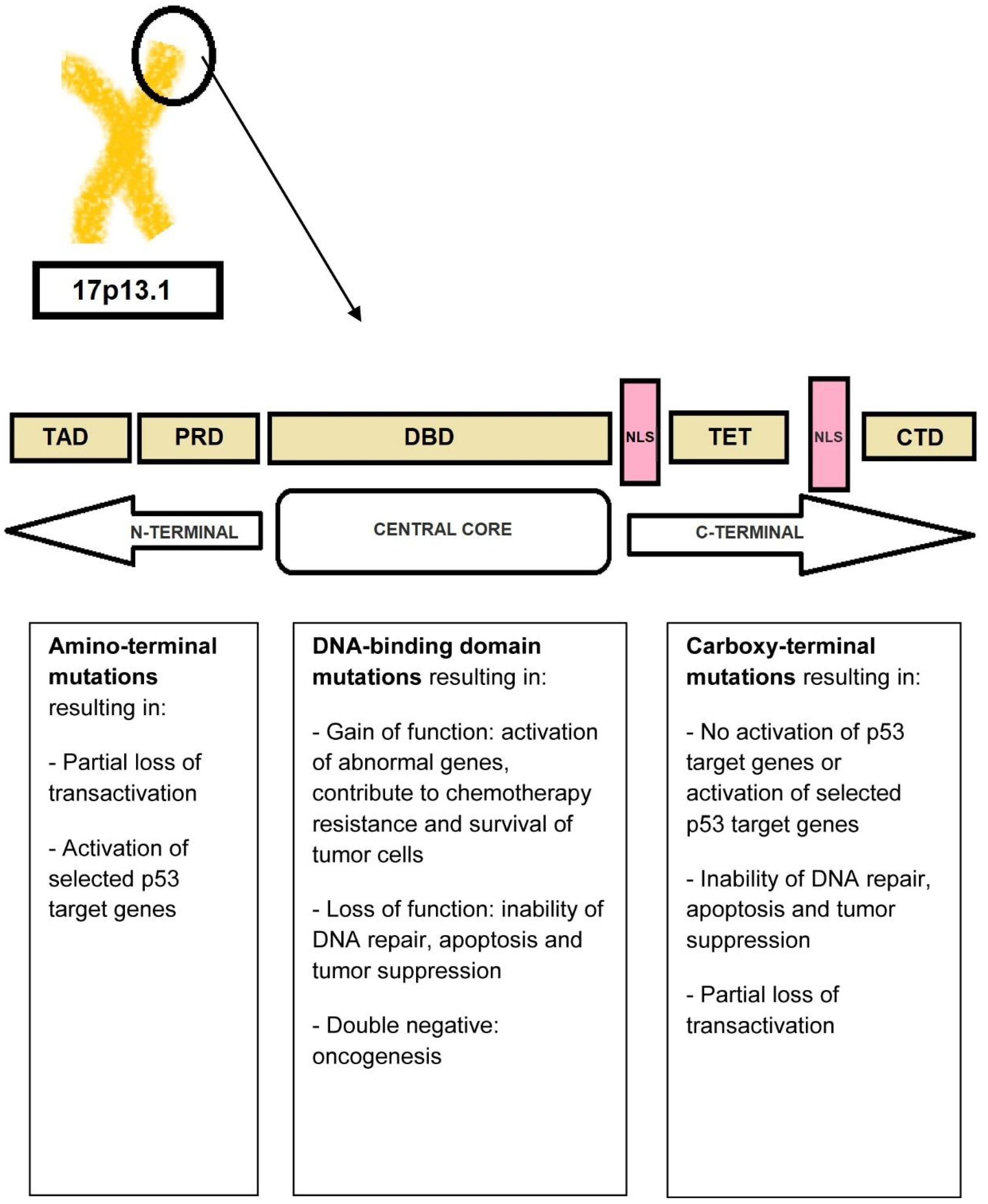
3.3. TP53 Mutations
3.4. Molecular Basis of t-AML
3.5. A Permissive Bone Marrow Microenvironment Facilitates t-AML Growth
3.6. Role of Pro-Inflammatory Cytokine Signaling
References
- Mcnerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527.
- Nilsson, C.; Linde, F.; Hulegårdh, E.; Garelius, H.; Lazarevic, V.; Antunovic, P.; Cammenga, J.; Deneberg, S.; Eriksson, A.; Jädersten, M.; et al. Characterization of therapy-related acute myeloid leukemia: Increasing incidence and prognostic implications. Haematologica 2022. online ahead of print.
- Fianchi, L.; Pagano, L.; Piciocchi, A.; Candoni, A.; Gaidano, G.; Breccia, M.; Criscuolo, M.; Specchia, G.; Pogliani, E.M.; Maurillo, L.; et al. Characteristics and outcome of therapy related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am. J. Hematol. 2015, 90, E80–E85.
- Kayser, S.; Döhner, K.; Jürgen Krauter, J.; Köhne, C.H.; Horst, H.A.; Held, G.; Lilienfeld-Toal, M.; Wilhelm, S.; Kündgen, A.; Götze, K.; et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145.
- Morton, L.M.; Dores, G.M.; Tucker, M.A.; Kim, C.J.; Onel, K.; Gilbert, E.S.; Fraumeni, J., Jr.; Curtis, R.E. Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975–2008. Blood 2013, 121, 2996–3004.
- Bhatia, S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin. Oncol. 2013, 40, 666–675.
- Jacoby, M.A.; De, R.; Pizarro, J.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014, 28, 1242–1251.
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376.
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S. The role of TP53 mutations in the origin and evolution of therapy-related AML. Nature 2015, 518, 552–555.
- Takahashi, K. Germline polymorphisms and the risk of therapy-related myeloid neoplasms. Best Pract. Res. Clin. Haematol. 2019, 32, 24–30.
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52.
- Abdelhameed, A.; Pond, G.R.; Mitsakakis, N.; Brandwein, J.; Chun, K.; Gupta, V.; Kamel-Reid, S.; Lipton, J.H.; Minden, M.D.; Schimmer, A.; et al. Outcome of patients who develop acute leukemia or myelodysplasia as a second malignancy after solid tumors treated surgically or with strategies that include chemotherapy and/or radiation. Cancer 2008, 112, 1513–1521.
- Loke, J.; Buka, R.; Craddock, C. Allogeneic stem cell transplantation for acute myeloid leukemia: Who, when, and how? Front. Immunol. 2021, 12, 659595.
- Marando, L.; Huntly, B.J.P. Molecular Landscape of Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. Curr. Oncol. Rep. 2020, 22, 61.
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2022, 171, 103607.
- Zhao, Q.; Ma, P.; Fu, P.; Wang, J.; Wang, K.; Chen, L.; Yang, Y. Myelodysplastic syndrome/acute myeloid leukemia following the use of poly-adp ribose polymerase (PARP) inhibitors: A real-world analysis of postmarketing surveillance data. Front. Pharmacol. 2022, 13, 912256.
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journey of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433.
- Heuser, M. Therapy-related myeloid neoplasms: Does knowing the origin help to guide treatment? Hematology 2016, 2016, 24–32.
- Sill, H.; Olipitz, W.; Zebisch, A.; Schulz, E.; Wölfler, A. Therapy-related myeloid neoplasms: Pathobiology and clinical characteristics. J. Pharmacol. 2011, 162, 792–805.
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398.
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377.
- Voso, M.T.; Falconi, G.; Fabiani, E. What’s new in the pathogenesis and treatment of therapy-related myeloid neoplasms. Blood 2021, 138, 749–757.
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085.
- Schratz, K.E.; Dezern, A.E. Genetic predisposition to myelodysplastic syndrome in clinical practice. Hematol. Oncol. Clin. N. Am. 2020, 34, 333–356.
- Churpek, J.E.; Marquez, R.; Neistadt, B.; Churpek, J.E.; Marquez, R.; Neistadt, B.; Claussen, K.; Lee, M.K.; Churpek, M.M.; Huo, D.; et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 2016, 122, 304–315.
- Schulz, E.; Valentin, A.; Ulz, P.; Beham-Schmid, C.; Lind, K.; Rupp, V.; Lackner, H.; Wölfler, A.; Zebisch, A.; Olipitz, W.; et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J. Med. Genet. 2012, 49, 422–428.
- Abd, E.l.; Wahab, N.; Shafik, N.F.; Shafik, R.E.; Taha, S.H.A.; Shafik, H.E.; Darwish, A.D. Association of CYP3A5*3 and CYP1A1*2C polymorphism with development of acute myeloid leukemia in Egyptian patients. Asian Pac. J. Cancer Prev. 2017, 18, 747–752.
- Bejar, R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017, 31, 1869–1871.
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, G.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105.
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604.
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16.
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074.
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089.
- Miller, M.; Shirole, N.; Tian, R.; Pal, D.; Sordella, R. The Evolution of TP53 Mutations: From Loss-of-Function to Separation-of-Function Mutants. J. Cancer Biol. Res. 2016, 4, 1091.
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878.
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286.
- Eliyahu, D.; Michalovitz, D.; Eliyahu, S.; Pinhasi-Kimhi, O.; Oren, M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 1989, 86, 8763–8767.
- Binsah, G.; Hagop, K.; Natalia, B.; Krocker, J.D.; Rios, A. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int. J. Mol. Sci. 2021, 22, 10782.
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614.
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211.
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165.
- Weinberg, O.K.; Siddon, A.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Dal Cin, P.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853.
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk Kooy, M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354.
- Naval, D.; Abhishek, M.; Tapan, K.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529.
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020, 26, 1549–1556.
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds, S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 2019, 51, 694–704.
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184.
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhne, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20.
- Sallman, D.A.; Mclemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823.
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Fu, B.; Tang, G.; Goswami, M.; Singh, R.; Kanagal-Shamanna, R.; Pierce, S.A.; et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk. Res. 2015, 39, 348–354.
- Caprioli, C.; Lussana, F.; Salmoiraghi, S.; Cavagna, R.; Buklijas, K.; Elidi, L.; Zanghi, P.; Michelato, A.; Delaini, F.; Oldani, E.; et al. Clinical significance of chromatinspliceosome acute myeloid leukemia: A report from the Northern Italy Leukemia Group (NILG) randomized trial 02/06. Haematologica 2021, 106, 2578–2587.
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk-stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622.
- Boddu, P.; Zeidan, A.M. Myeloid disorders after autoimmune disease. Best Pract. Res. Clin. Haematol. 2019, 32, 74–88.
- Daniel, V.; Trojan, K.; Opelz, G. Immunosuppressive drugs affect induction of IFNy+ Treg in vitro. Hum. Immunol. 2016, 77, 146–152.
- Ustun, C.; Miller, J.S.; Munn, D.H.; Weisdorf, D.J.; Blazar, B.R. Regulatory T cells in acute myelogenous leukemia: Is it time for immunomodulation? Blood 2011, 118, 5084–5095.
- Wang, M.; Zhang, C.; Tian, T.; Zhang, T.; Wang, R.; Han, F.; Zhong, C.; Hua, M.; Ma, D. Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)-α-TNF receptor-2 pathway. Front. Immunol. 2018, 9, 1274.
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2009, 23, 510–518.
- Bifano Pimenta, D.; Araujo Varela, V.; Santos Datoguia, T.; Caraciolo, V.B.; Lopes, G.H.; Pereira, W.O. The bone marrow microenvironment mechanisms in acute myeloid leukemia. Front. Cell Dev. Biol. 2021, 9, 764698.
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Turzanski, J.; Grundy, M.; Russell, N.H.; Pallis, M. Interleukin-1beta maintains an apoptosis-resistant phenotype in the blast cells of acute myeloid leukaemia via multiple pathways. Leukemia 2004, 18, 1662–1670.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Delwel, R.; Van Buitenen, C.; Salem, M.; Bot, F.; Gillis, S.; Kaushansky, K.; Altrock, B.; Löwenberg, B. Lowenberg, Interleukin-1 stimulates proliferation of acute myeloblastic leukemia cells by induction of granulocytemacrophage colony-stimulating factor release. Blood 1989, 74, 586–593.
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992, 356, 768–774.
- Alonso-Alvarez, S.; Magnano, L.; Alcoceba, M.; Andrade-Campos, M.; Espinosa-Lara, N.; Rodríguez, G.; Mercadal, S.; Carro, I.; Sancho, J.M.; Moreno, M.; et al. Risk of, and survival following, histological transformation in follicular lymphoma in the rituximab era. A retrospective multicentre study by the Spanish GELTAMO group. Br. J. Haematol. 2017, 178, 699–708.
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315.
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15.
- Katsumura, K.R.; Ong, I.M.; DeVilbiss, A.W.; Sanalkumar, R.; Bresnick, E.H. GATA factor-dependent positivefeedback circuit in acute myeloid leukemia cells. Cell Rep. 2016, 16, 2428–2441.
- Carey, A.; Edwards, D.K.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 2017, 18, 3204–3218.
- Asano, Y.; Shibata, S.; Kobayashi, S.; Okamura, S.; Niho, Y. Interleukin-10 inhibits the autocrine growth of leukemic blast cells from patients with acute myeloblastic leukemia. Int. J. Hematol. 1997, 66, 445–450.
- Tsimberidou, A.M.; Estey, E.; Wen, S.; Pierce, S.; Kantarjian, H.; Albitar, M.; Kurzrock, R. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer 2008, 113, 1605–1613.
- Yao, C.J.; Du, W.; Chen, H.B.; Xiao, S.; Wang, C.H.; Fan, Z.L. Associations of IL-10 gene polymorphisms with acute myeloid leukemia in Hunan, China. Asian Pac. J. Cancer Prev. 2013, 14, 2439–2442.

