Therapy-related acute myeloid leukemia (t-AML) is one of the most serious long-term complications of cancer chemotherapy. Various cytotoxic agents and exposure to ionizing radiation can lead to the development of t-AML, which is usually associated with adverse genetic changes and a poor prognosis.
- therapy-related
- acute myeloid leukemia
- AML
1. Introduction
2. Etiology of t-AML
The occurrence of t-AML is typically a late adverse effect after the administration of a leukemogenic agent. The main causative agents that have been linked to the development of t-AML include alkylating agents and topoisomerase II inhibitors. Other factors causally associated with t-AML include antimetabolites, anthracyclines (through their ability to inhibit DNA topoisomerases I and II), antimicrotubule (antitubulin) agents (usually in combination with other cytotoxic agents), and ionizing radiation [16][15]. Moreover, recent studies have reported an increased incidence of MDS and AML after the administration of targeted treatment with the poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) inhibitors olaparib, niraparib, and rucaparib for metastatic or recurrent epithelial ovarian cancer, particularly in women with germline mutations in BRCA1 and BRCA2 (mBRCA) genes [17][16]. The cytotoxic factors associated with t-AML are shown in Table 1.|
Drug Class |
Mechanism of Action |
|---|---|
|
Alkylating Agents Cyclophosphamide, * cisplatin, carboplatin, melphalan, busulphan, chlorambucil, lomustin, carmustine, dacarbazine, procarbazine, thiotepa, mitomycin C |
Creation of bonds in one or both DNA strands, through alkylation |
|
Topoisomerase II Inhibitors Etoposide, teniposide, doxorubicin, idarubicin, daunorubicin, mitoxantrone, * actinomycin D, amsacrine |
“Topoisomerase II poisons” convert topoisomerase II into a DNA-damaging enzyme |
|
Antimetabolites Fludarabine, cladribine, * pentostatin, thiopurines (6-mercaptopurine, * 6-thioguanine, azathioprine *), mycophenolate mofetil * |
They act as mimics of other molecules, and in this way, they interfere with DNA and RNA synthesis |
|
Antitubulin Agents Vinblastine, vindesine, vincristine, docetaxel, paclitaxel |
Antimitotic agents that bind tubulin dimers and disrupt the formation of mitotic spindle |
|
Poly (ADP-Ribose) Polymerase (PARP) Inhibitors Olaparib, talazoparib, niraparib, rucaparib |
Inhibitors of the PARP family of enzymes inhibit homologous recombination repair (PARP enzymes, activated by DNA damage, repair the single-helix DNA breaks by forming branched PAR chains that serve as a docking platform for DNA repair enzymes) |
Abbreviations: t-AML, therapy-related acute myeloid leukemia; PARP, poly (ADP-ribose) polymerase. * Widely used as immunosuppressive agents in the treatment of autoimmune and immune-mediated inflammatory disorders.
Types of t-AML
2.1. Types of t-AML
The most common type of t-AML occurs 4–10 years after exposure to alkylating agents and/or ionizing radiation. The mechanism of action of alkylating agents depends on the creation of bonds, through alkylation, in one or both strands of the DNA double helix [18][17]. The risk of this complication peaks 5–10 years after the start of chemotherapy. These patients frequently present with MDS, which may then progress to frank AML [19][18]. This subtype of t-AML is often associated with a loss of genetic material, typically deletions of chromosomes 5 and 7. It is also commonly associated with chromosome 17 or 17p deletion, complex karyotype, and TP53 mutations [8,16][8][15]. A second distinct subtype of t-AML accounting for 20–30% of t-AML has been identified as a complication of treatment with topoisomerase II inhibitors (also called “topoisomerase II poisons”), such as epipodophyllotoxins. In contrast to alkylating-agent-related AML, topoisomerase-inhibitor-related AML develops after a relatively short latency period (1–5 years) and is not preceded by MDS [20][19]. Epipodophyllotoxins block cells in the late S to G2 phase of the cell cycle. Their major target is the enzyme DNA topoisomerase IIA, a nuclear enzyme that is essential in DNA replication by creating double-stranded cuts in DNA. The binding of epipodophyllotoxins to the enzyme–DNA complex results in persistence of the transient, cleavable form of the complex and, thus, renders it susceptible to irreversible double-strand breaks [21][20]. Exposure to drugs that inhibit topoisomerase II―i.e., the epipodophyllotoxins etoposide and teniposide; the anthracyclines daunorubicin, doxorubicin, and epirubicin; and the anthracenedione mitoxantrone―predisposes patients to the development of t-AML with balanced chromosomal translocations, including KMT2A (MLL) translocations at chromosome band 11q23, t(8;21), t(16;16), t(15;17), and t(9;22), and NUP98 translocations at chromosome band 11p15.5 [15,22][21][22].3. Pathophysiology of t-AML
3.1. Genetic Predisposition for t-AML
Although many patients are exposed to mutagenic agents as part of their treatment, only a small minority of them develop t-AML in their lifetime. Additionally, among individuals exposed to the same amount of cytotoxic therapy, only a few develop t-AML. This suggests that genetic predisposition may be a key factor. Clinicians should be aware that as many as 20% of otherwise typical cases of t-AML occurring after treatment for breast or ovarian cancer may actually be AML with germline predisposition caused by inherited mutations in the DNA repair genes (BRCA1, BRCA2, PALB2, TP53, or CHEK2), typical of familial cancer predisposition syndromes [23,24,25,26][23][24][25][26]. Somatic (acquired) mutations in TP53 have been detected in most human cancers including breast cancer. However, inherited mutations (transmitted through the germline) of TP53 also underlie the Li–Fraumeni syndrome, a rare familial association of breast cancer in young women, leukemia (AML/MDS), childhood sarcomas (“BLS” syndrome), and/or other neoplasms, which is transmitted as an autosomal dominant trait. For example, a woman who has survived breast carcinoma and/or sarcoma and is now facing t-AML with del(17) on karyotype and TP53 mutation on next-generation sequencing (NGS) should be screened for Li–Fraumeni syndrome (even if her family history is not indicative of it) [22]. Knowing that an important subgroup of younger patients with t-AML carry germline mutations in cancer predisposition genes is important not only for their treatment (e.g., selection of appropriate family donors for allogeneic HCT) but also for identifying family members who may be at high risk for the development of tumors. Studies have also examined whether polymorphisms in genes involved in the metabolism of alkylating agents and topoisomerase II inhibitors such as cytochrome P450 enzymes (CYP3A4/CYP3A5), GSTM1, GSTT1, and NQO1 may confer an increased risk of t-AML, with so far controversial results [10,27][10][27].3.2. Current Model for the Molecular Pathogenesis of t-AML
Beyond genetic predisposition and the inheritance of familial cancer genes, NGS has revealed that, in many patients, the first step in the process of t-AML development is age-related clonal hematopoiesis also known as clonal hematopoiesis of indeterminate potential (CHIP) [28]. According to this model, HSC clones harboring somatic TP53 or PPM1D mutations are detected in patients before chemotherapy exposure [29,30][29][30]. Owing to its competitive advantage, the TP53-mutant clone enlarges in the bone marrow after chemotherapy administered for a neoplastic or non-neoplastic disorder. The acquisition of additional mutations or the emergence of cytogenetic abnormalities such as chromosome 5/5q or 7/7q deletion leads to a selection of subclones of hematopoietic cells with an increasingly impaired differentiation capacity, which drives leukemic transformation (Figure 1) [8,22,31][8][22][31].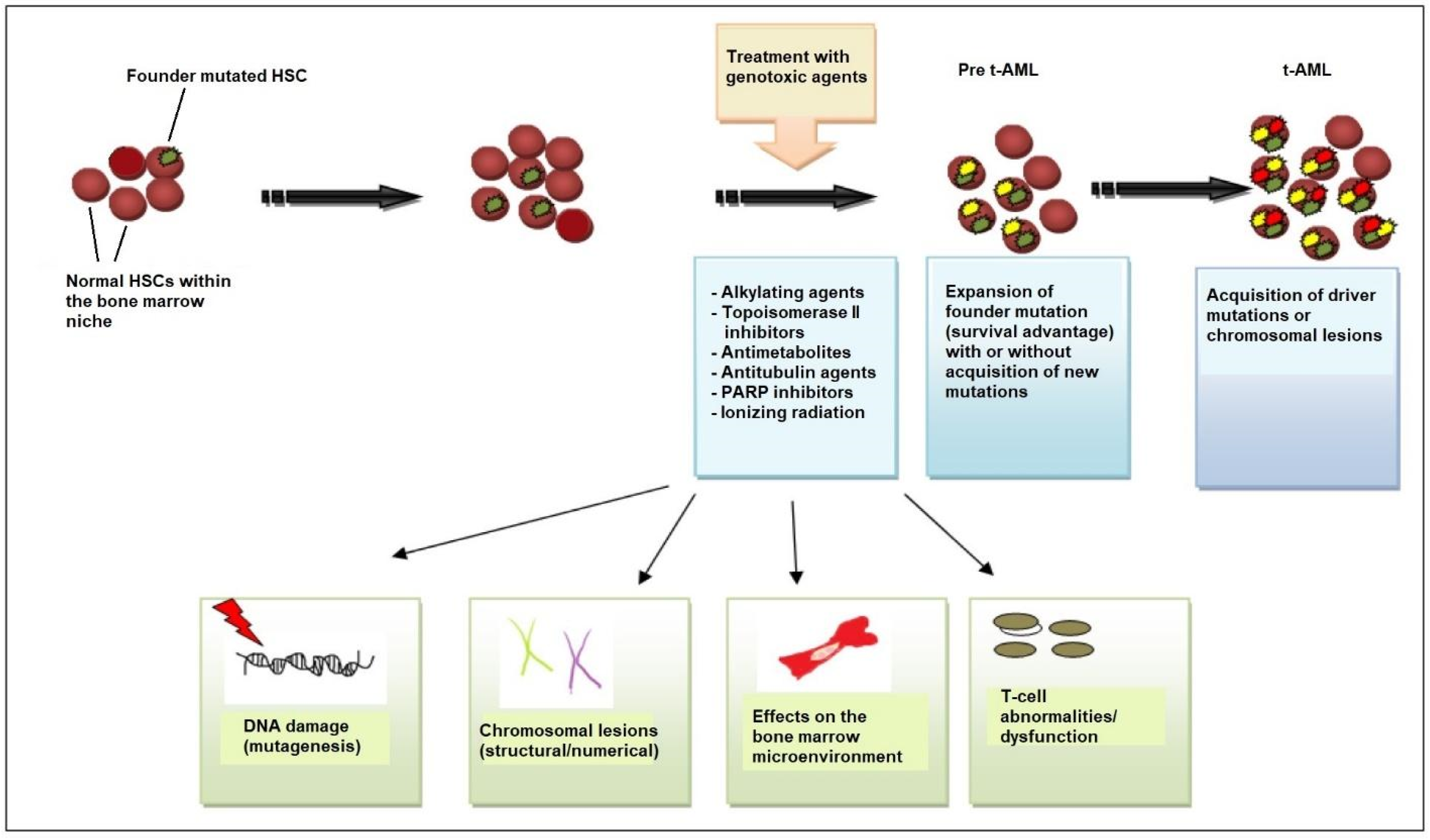
3.3. TP53 Mutations
The TP53 tumor-suppressor gene, located on the short arm of chromosome 17, encodes a 53-kd nuclear phosphoprotein that suppresses cell growth in response to DNA damage through several mechanisms including cell-cycle arrest at the G1/S checkpoint, activation of DNA-repair enzymes, and initiation of apoptosis in cases of severe DNA damage [9]. From a mechanistic point of view, the activation of the DNA damage checkpoint results in the formation of TP53 homo-tetramers (tetramerization is essential for p53 activation in vivo). Activated TP53 then interacts with other tumor suppressors such as p21Waf1/Cip1, p63, and p73, activates numerous microRNAs (including the miR-34 family of miRNAs) and proapoptotic proteins (BCL-XL, BCL2, Bax), and alters the function of mTOR kinase. The transcriptional activity of TP53 is carried out by five distinct domains including the transactivation activation domain (TAD) and the proline-rich domain (PRD), located at the N-terminal portion of the p53 protein, the core DNA-binding domain (DBD) and the tetramerization domain (TET) (responsible for the oligomerization of the p53 protein, which exists as a tetramer), and the carboxy-terminal regulatory domain (CTD) at the C-terminal portion of the protein. The C-terminal portion also includes several nuclear localization sequences (NLS) (Figure 2) [34,35][34][35].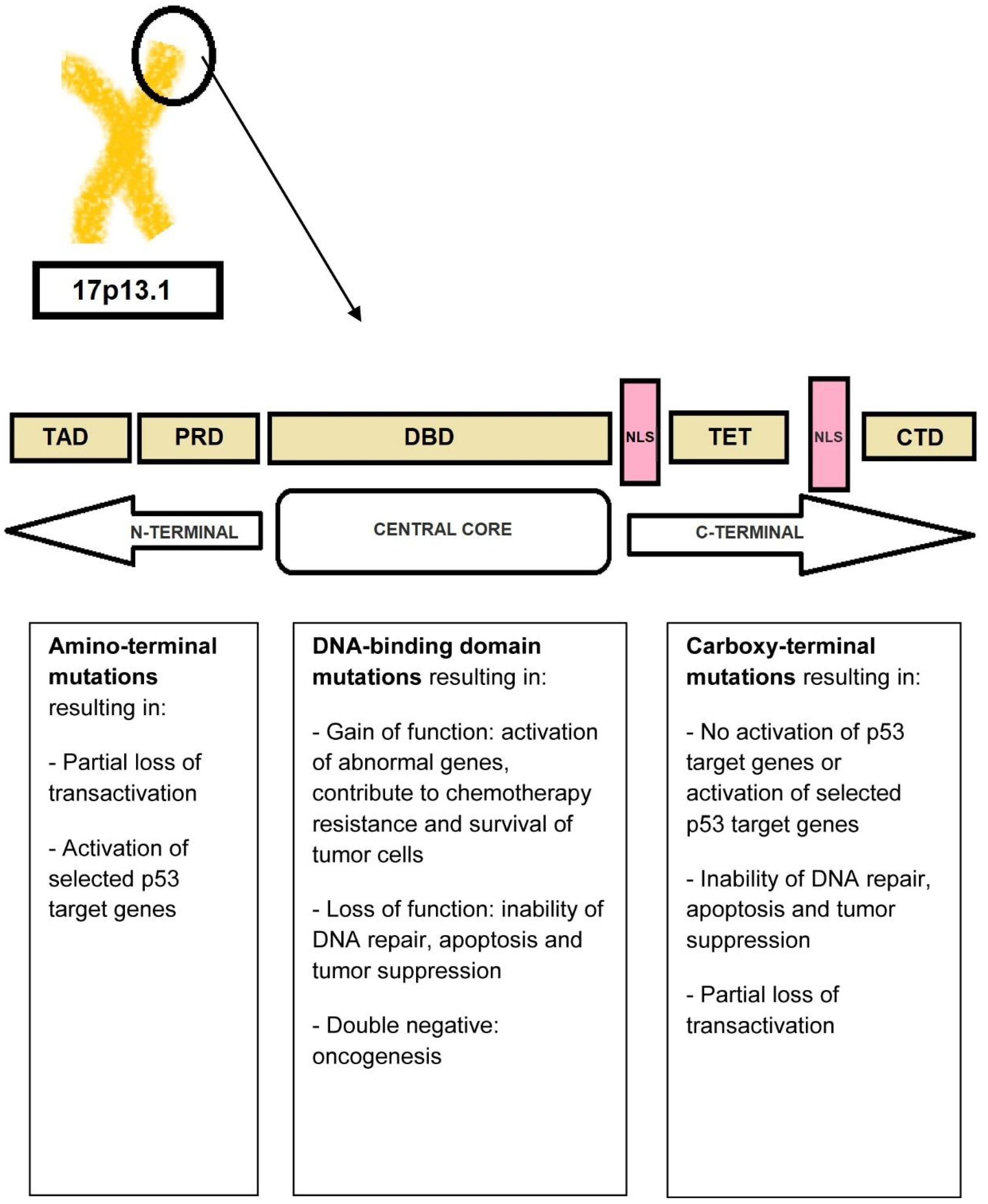
3.4. Molecular Basis of t-AML
Lindsley and coworkers investigated the genetic basis of t-AML and secondary AML (s-AML) and identified three distinct subtypes of somatic mutations: (i) secondary-type mutations involving eight genes (SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, and STAG2) which are commonly linked to MDS and are now recognized as “AML with myelodysplasia-related (MR) gene mutation” in 2022 ELN recommendations; (ii) TP53 mutations, which are associated with complex karyotypes (often monosomal, with frequent abnormalities of chromosomes 5 and 7), intrinsic therapy resistance, and very poor survival; and (iii) “de novo”-type (or pan-AML-type) mutations including NPM1 mutations, KMT2A (MLL) rearrangements at 11q23 locus, core-binding-factor (CBF) chromosomal rearrangements, myeloid-transcription-factor mutations (e.g., RUNX1, CEBPA, and GATA2), signal-transduction-protein mutations (e.g., FLT3, N-RAS, and K-RAS), and other mutations (e.g., IDH1, IDH2, and WT1) [8]. “Secondary-type” mutations are found in 30% of t-AML cases and are associated with poor outcomes. TP53 mutations are seen in ~50% of t-AML cases. NPM1 mutations are identified in 5.4% of patients without concurrent “secondary-type” or TP53 mutations [8[8][30][52],30,52], suggesting that there is a non-random pattern of co-mutations with mutual exclusivity between TP53 and NPM1 mutations in t-AML. Patients with t-AML with “secondary-type” mutations are significantly older than patients with “de novo”/pan-AML mutations. Clinically, t-AML with “secondary-type” mutations closely resembles s-AML. In contrast, patients with t-AML with “de novo”-type mutations closely resemble patients with primary de novo AML [8,53][8][53]. Recently, Papaemmanuil and coworkers presented another categorization of patients with AML into 16 distinct molecular subgroups [54]. Regarding t-AML, the majority of patients had high relapse rates and poor prognosis, regardless of the achievement of early minimal residual disease (MRD) negativity. However, beyond this general rule, patients with t-AML who achieved CR, including those with TP53 mutations, seemed to benefit from HCT. Additionally, patients who had >2 mutations had worse prognosis compared to those who carried a single-gene mutation. The prognosis was even worse in patients with TP53 mutations or inv(3)/t(3;3), resulting in deregulated MECOM (EVI1) and GATA2 expression [54]. However, it should be emphasized that even patients carrying TP53 mutations benefited from HCT, especially in CR1 (and less so after CR2) [54]. Clearly, the past decade has reshaped our the view of t-AML. Rather than considering t-AML as one clinical entity, it is more appropriate to view t-AML as at least three molecular types (according to the Lindsley model) that vary in prevalence with age, each bearing more similarity to AML with the same genetic alterations and no leukemogenic exposure.3.5. A Permissive Bone Marrow Microenvironment Facilitates t-AML Growth
The immune system inside the bone marrow is also involved in the development of t-AML. Population-based studies show that AML is more common among patients with autoimmune diseases than the general population [55,56][55][56]. The risk of developing t-AML appears to be related to the type of autoimmune disease and type and duration of its treatment. Drugs used to treat autoimmune diseases such as azathioprine, mitoxantrone, and cyclophosphamide may directly damage DNA and increase the risk of leukemogenesis [55,56][55][56]. However, drugs used in autoimmune disorders such as azathioprine may also affect the balance between T-cell subsets. Experiments in mice have shown that high doses of azathioprine downregulate regulatory T-cells (T-regs), whereas lower doses upregulate T-regs [57]. Abnormalities in T-regs may play a role in the transition of MDS to AML [58,59][58][59]. For example, in patients with clinically stable MDS (regardless of the disease stage), T-reg levels remain stable, but upon transformation to AML, an increase in T-reg numbers is noted both in the marrow and in the peripheral blood [60]. In addition, reduced immune surveillance by cytotoxic T-cells is seen in patients with autoimmune disorders. Chronic inflammatory signaling and inflammatory conditions can modulate the bone marrow microenvironment and facilitate the survival and proliferation of leukemic cells [56,61][56][61]. Notably, the transcription factor nuclear factor-κB (NF-κB), a central pro-inflammatory mediator, and polymorphisms in the interleukin-1 receptor antagonist (IL-1Rα) are involved in both autoimmune diseases and leukemogenesis [56,62,63,64,65][56][62][63][64][65].3.6. Role of Pro-Inflammatory Cytokine Signaling
The response of a body to a cancer is not a unique mechanism but has many parallels with the chronic inflammation seen in chronic infections. Balkwill and Mantovani’s metaphor, stating that if genetic damage is “the match that lights the fire of cancer”, some types of inflammation provide “the fuel that feeds the flames”, puts emphasis on inflammation as a major contributor in the growth of some cancers [66]. Multifaceted activation of the immune system and chronic inflammation accompany many hematologic neoplasms including MDS and AML. Certain pro-inflammatory cytokines such as the tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6) play an important role in the development of AML. IL-1 occurs as two structurally related polypeptides (IL-1α and IL-1β), each of which has a broad spectrum of both beneficial and harmful biologic effects. IL-1β, in particular, contributes to the proliferation and survival of leukemic cells [65,67][65][67]. It has been found that IL-1β participates in the immune response through a dual mechanism: the activation of the IL-1 receptor (IL-1R)/Toll-like receptor (TLR) and caspase-1 activation [68,69][68][69]. The activation of IL-1β induces a signaling cascade that leads to the phosphorylation and ubiquitination of MyD88, IRAK-4, and TRAF-6 and, ultimately, NF-κB activation [70]. At the same time, IL-1β induces the activation of p38 mitogen-activated protein kinase (MAPK), as well as the activation of the transcription factor GATA2 [70,71,72,73][70][71][72][73]. Both p38 MAPK and GATA2 contribute to the proliferation of leukemic blasts. Monocytes and macrophages represent the main cell source of IL-1β. IL-1β may be a target for future therapies in AML. IL-35, transforming growth factor β (TGF-β), and IL-10 have also been implicated in the pathogenesis of AML. IL-35 belongs to the IL-12 family and is produced by T-regs. In patients with AML, high concentrations of IL-35 have been found in bone marrow plasma, corresponding to an increased proportion of T-regs [74,75,76][74][75][76]. TGF-β contributes to many cellular processes including cell survival, proliferation, and migration. In AML, TGF-β inhibits the proliferation of leukemic stem cells (LSCs), maintaining their longevity. The administration of DNA damaging agents, in association with disturbances in T-cell subsets and pro-inflammatory changes in the bone marrow microenvironment, may favor the development of t-AML in patients with autoimmune disorders.References
- Mcnerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527.
- Nilsson, C.; Linde, F.; Hulegårdh, E.; Garelius, H.; Lazarevic, V.; Antunovic, P.; Cammenga, J.; Deneberg, S.; Eriksson, A.; Jädersten, M.; et al. Characterization of therapy-related acute myeloid leukemia: Increasing incidence and prognostic implications. Haematologica 2022. online ahead of print.
- Fianchi, L.; Pagano, L.; Piciocchi, A.; Candoni, A.; Gaidano, G.; Breccia, M.; Criscuolo, M.; Specchia, G.; Pogliani, E.M.; Maurillo, L.; et al. Characteristics and outcome of therapy related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am. J. Hematol. 2015, 90, E80–E85.
- Kayser, S.; Döhner, K.; Jürgen Krauter, J.; Köhne, C.H.; Horst, H.A.; Held, G.; Lilienfeld-Toal, M.; Wilhelm, S.; Kündgen, A.; Götze, K.; et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145.
- Morton, L.M.; Dores, G.M.; Tucker, M.A.; Kim, C.J.; Onel, K.; Gilbert, E.S.; Fraumeni, J., Jr.; Curtis, R.E. Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975–2008. Blood 2013, 121, 2996–3004.
- Bhatia, S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin. Oncol. 2013, 40, 666–675.
- Jacoby, M.A.; De, R.; Pizarro, J.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014, 28, 1242–1251.
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376.
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S. The role of TP53 mutations in the origin and evolution of therapy-related AML. Nature 2015, 518, 552–555.
- Takahashi, K. Germline polymorphisms and the risk of therapy-related myeloid neoplasms. Best Pract. Res. Clin. Haematol. 2019, 32, 24–30.
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52.
- Abdelhameed, A.; Pond, G.R.; Mitsakakis, N.; Brandwein, J.; Chun, K.; Gupta, V.; Kamel-Reid, S.; Lipton, J.H.; Minden, M.D.; Schimmer, A.; et al. Outcome of patients who develop acute leukemia or myelodysplasia as a second malignancy after solid tumors treated surgically or with strategies that include chemotherapy and/or radiation. Cancer 2008, 112, 1513–1521.
- Loke, J.; Buka, R.; Craddock, C. Allogeneic stem cell transplantation for acute myeloid leukemia: Who, when, and how? Front. Immunol. 2021, 12, 659595.
- Marando, L.; Huntly, B.J.P. Molecular Landscape of Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. Curr. Oncol. Rep. 2020, 22, 61.
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2022, 171, 103607.
- Zhao, Q.; Ma, P.; Fu, P.; Wang, J.; Wang, K.; Chen, L.; Yang, Y. Myelodysplastic syndrome/acute myeloid leukemia following the use of poly-adp ribose polymerase (PARP) inhibitors: A real-world analysis of postmarketing surveillance data. Front. Pharmacol. 2022, 13, 912256.
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journey of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433.
- Heuser, M. Therapy-related myeloid neoplasms: Does knowing the origin help to guide treatment? Hematology 2016, 2016, 24–32.
- Sill, H.; Olipitz, W.; Zebisch, A.; Schulz, E.; Wölfler, A. Therapy-related myeloid neoplasms: Pathobiology and clinical characteristics. J. Pharmacol. 2011, 162, 792–805.
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398.
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377.
- Voso, M.T.; Falconi, G.; Fabiani, E. What’s new in the pathogenesis and treatment of therapy-related myeloid neoplasms. Blood 2021, 138, 749–757.
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085.
- Schratz, K.E.; Dezern, A.E. Genetic predisposition to myelodysplastic syndrome in clinical practice. Hematol. Oncol. Clin. N. Am. 2020, 34, 333–356.
- Churpek, J.E.; Marquez, R.; Neistadt, B.; Churpek, J.E.; Marquez, R.; Neistadt, B.; Claussen, K.; Lee, M.K.; Churpek, M.M.; Huo, D.; et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 2016, 122, 304–315.
- Schulz, E.; Valentin, A.; Ulz, P.; Beham-Schmid, C.; Lind, K.; Rupp, V.; Lackner, H.; Wölfler, A.; Zebisch, A.; Olipitz, W.; et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J. Med. Genet. 2012, 49, 422–428.
- Abd, E.l.; Wahab, N.; Shafik, N.F.; Shafik, R.E.; Taha, S.H.A.; Shafik, H.E.; Darwish, A.D. Association of CYP3A5*3 and CYP1A1*2C polymorphism with development of acute myeloid leukemia in Egyptian patients. Asian Pac. J. Cancer Prev. 2017, 18, 747–752.
- Bejar, R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017, 31, 1869–1871.
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, G.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105.
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604.
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16.
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074.
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089.
- Miller, M.; Shirole, N.; Tian, R.; Pal, D.; Sordella, R. The Evolution of TP53 Mutations: From Loss-of-Function to Separation-of-Function Mutants. J. Cancer Biol. Res. 2016, 4, 1091.
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878.
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286.
- Eliyahu, D.; Michalovitz, D.; Eliyahu, S.; Pinhasi-Kimhi, O.; Oren, M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 1989, 86, 8763–8767.
- Binsah, G.; Hagop, K.; Natalia, B.; Krocker, J.D.; Rios, A. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int. J. Mol. Sci. 2021, 22, 10782.
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614.
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211.
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165.
- Weinberg, O.K.; Siddon, A.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Dal Cin, P.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853.
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk Kooy, M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354.
- Naval, D.; Abhishek, M.; Tapan, K.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529.
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020, 26, 1549–1556.
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds, S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 2019, 51, 694–704.
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184.
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhne, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20.
- Sallman, D.A.; Mclemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823.
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Fu, B.; Tang, G.; Goswami, M.; Singh, R.; Kanagal-Shamanna, R.; Pierce, S.A.; et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk. Res. 2015, 39, 348–354.
- Caprioli, C.; Lussana, F.; Salmoiraghi, S.; Cavagna, R.; Buklijas, K.; Elidi, L.; Zanghi, P.; Michelato, A.; Delaini, F.; Oldani, E.; et al. Clinical significance of chromatinspliceosome acute myeloid leukemia: A report from the Northern Italy Leukemia Group (NILG) randomized trial 02/06. Haematologica 2021, 106, 2578–2587.
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk-stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622.
- Boddu, P.; Zeidan, A.M. Myeloid disorders after autoimmune disease. Best Pract. Res. Clin. Haematol. 2019, 32, 74–88.
- Daniel, V.; Trojan, K.; Opelz, G. Immunosuppressive drugs affect induction of IFNy+ Treg in vitro. Hum. Immunol. 2016, 77, 146–152.
- Ustun, C.; Miller, J.S.; Munn, D.H.; Weisdorf, D.J.; Blazar, B.R. Regulatory T cells in acute myelogenous leukemia: Is it time for immunomodulation? Blood 2011, 118, 5084–5095.
- Wang, M.; Zhang, C.; Tian, T.; Zhang, T.; Wang, R.; Han, F.; Zhong, C.; Hua, M.; Ma, D. Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)-α-TNF receptor-2 pathway. Front. Immunol. 2018, 9, 1274.
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2009, 23, 510–518.
- Bifano Pimenta, D.; Araujo Varela, V.; Santos Datoguia, T.; Caraciolo, V.B.; Lopes, G.H.; Pereira, W.O. The bone marrow microenvironment mechanisms in acute myeloid leukemia. Front. Cell Dev. Biol. 2021, 9, 764698.
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Turzanski, J.; Grundy, M.; Russell, N.H.; Pallis, M. Interleukin-1beta maintains an apoptosis-resistant phenotype in the blast cells of acute myeloid leukaemia via multiple pathways. Leukemia 2004, 18, 1662–1670.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Delwel, R.; Van Buitenen, C.; Salem, M.; Bot, F.; Gillis, S.; Kaushansky, K.; Altrock, B.; Löwenberg, B. Lowenberg, Interleukin-1 stimulates proliferation of acute myeloblastic leukemia cells by induction of granulocytemacrophage colony-stimulating factor release. Blood 1989, 74, 586–593.
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992, 356, 768–774.
- Alonso-Alvarez, S.; Magnano, L.; Alcoceba, M.; Andrade-Campos, M.; Espinosa-Lara, N.; Rodríguez, G.; Mercadal, S.; Carro, I.; Sancho, J.M.; Moreno, M.; et al. Risk of, and survival following, histological transformation in follicular lymphoma in the rituximab era. A retrospective multicentre study by the Spanish GELTAMO group. Br. J. Haematol. 2017, 178, 699–708.
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315.
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15.
- Katsumura, K.R.; Ong, I.M.; DeVilbiss, A.W.; Sanalkumar, R.; Bresnick, E.H. GATA factor-dependent positivefeedback circuit in acute myeloid leukemia cells. Cell Rep. 2016, 16, 2428–2441.
- Carey, A.; Edwards, D.K.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 2017, 18, 3204–3218.
- Asano, Y.; Shibata, S.; Kobayashi, S.; Okamura, S.; Niho, Y. Interleukin-10 inhibits the autocrine growth of leukemic blast cells from patients with acute myeloblastic leukemia. Int. J. Hematol. 1997, 66, 445–450.
- Tsimberidou, A.M.; Estey, E.; Wen, S.; Pierce, S.; Kantarjian, H.; Albitar, M.; Kurzrock, R. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer 2008, 113, 1605–1613.
- Yao, C.J.; Du, W.; Chen, H.B.; Xiao, S.; Wang, C.H.; Fan, Z.L. Associations of IL-10 gene polymorphisms with acute myeloid leukemia in Hunan, China. Asian Pac. J. Cancer Prev. 2013, 14, 2439–2442.