Overall, the prognosis for patients with t-AML is considerably worse than that for patients with primary de novo AML. It is estimated that the median overall survival (OS) is 8–10 months and the 5-year OS is 10–20% [
4,
10,
11,
12]. Therefore, allogeneic hematopoietic-cell transplantation (HCT) from a suitable donor has been established practice for all patients with non-acute-promyelocytic-leukemia (non-APL) t-AML in first complete remission (CR1) for more than 30 years [
13]. More recently, however, this generalized approach has become controversial due to the increasing recognition of the prognostic significance of the molecular tumor genetics in relation to the risk of relapse [
14].
2. Etiology of t-AML
The occurrence of t-AML is typically a late adverse effect after the administration of a leukemogenic agent. The main causative agents that have been linked to the development of t-AML include alkylating agents and topoisomerase II inhibitors. Other factors causally associated with t-AML include antimetabolites, anthracyclines (through their ability to inhibit DNA topoisomerases I and II), antimicrotubule (antitubulin) agents (usually in combination with other cytotoxic agents), and ionizing radiation [
16]. Moreover, recent studies have reported an increased incidence of MDS and AML after the administration of targeted treatment with the poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) inhibitors olaparib, niraparib, and rucaparib for metastatic or recurrent epithelial ovarian cancer, particularly in women with germline mutations in
BRCA1 and
BRCA2 (
mBRCA) genes [
17]. The cytotoxic factors associated with t-AML are shown in
Table 1.
Table 1. Chemotherapeutic agents implicated in t-AML.
Abbreviations: t-AML, therapy-related acute myeloid leukemia; PARP, poly (ADP-ribose) polymerase. * Widely used as immunosuppressive agents in the treatment of autoimmune and immune-mediated inflammatory disorders.
Types of t-AML
The most common type of t-AML occurs 4–10 years after exposure to alkylating agents and/or ionizing radiation. The mechanism of action of alkylating agents depends on the creation of bonds, through alkylation, in one or both strands of the DNA double helix [
18]. The risk of this complication peaks 5–10 years after the start of chemotherapy. These patients frequently present with MDS, which may then progress to frank AML [
19]. This subtype of t-AML is often associated with a loss of genetic material, typically deletions of chromosomes 5 and 7. It is also commonly associated with chromosome 17 or 17p deletion, complex karyotype, and
TP53 mutations [
8,
16].
A second distinct subtype of t-AML accounting for 20–30% of t-AML has been identified as a complication of treatment with topoisomerase II inhibitors (also called “topoisomerase II poisons”), such as epipodophyllotoxins. In contrast to alkylating-agent-related AML, topoisomerase-inhibitor-related AML develops after a relatively short latency period (1–5 years) and is not preceded by MDS [
20]. Epipodophyllotoxins block cells in the late S to G2 phase of the cell cycle. Their major target is the enzyme DNA topoisomerase IIA, a nuclear enzyme that is essential in DNA replication by creating double-stranded cuts in DNA. The binding of epipodophyllotoxins to the enzyme–DNA complex results in persistence of the transient, cleavable form of the complex and, thus, renders it susceptible to irreversible double-strand breaks [
21]. Exposure to drugs that inhibit topoisomerase II―i.e., the epipodophyllotoxins etoposide and teniposide; the anthracyclines daunorubicin, doxorubicin, and epirubicin; and the anthracenedione mitoxantrone―predisposes patients to the development of t-AML with balanced chromosomal translocations, including
KMT2A (
MLL) translocations at chromosome band 11q23, t(8;21), t(16;16), t(15;17), and t(9;22), and
NUP98 translocations at chromosome band 11p15.5 [
15,
22].
3. Pathophysiology of t-AML
3.1. Genetic Predisposition for t-AML
Although many patients are exposed to mutagenic agents as part of their treatment, only a small minority of them develop t-AML in their lifetime. Additionally, among individuals exposed to the same amount of cytotoxic therapy, only a few develop t-AML. This suggests that genetic predisposition may be a key factor.
Clinicians should be aware that as many as 20% of otherwise typical cases of t-AML occurring after treatment for breast or ovarian cancer may actually be AML with germline predisposition caused by inherited mutations in the DNA repair genes (
BRCA1,
BRCA2,
PALB2,
TP53, or
CHEK2), typical of familial cancer predisposition syndromes [
23,
24,
25,
26]. Somatic (acquired) mutations in
TP53 have been detected in most human cancers including breast cancer. However, inherited mutations (transmitted through the germline) of
TP53 also underlie the Li–Fraumeni syndrome, a rare familial association of breast cancer in young women, leukemia (AML/MDS), childhood sarcomas (“BLS” syndrome), and/or other neoplasms, which is transmitted as an autosomal dominant trait. For example, a woman who has survived breast carcinoma and/or sarcoma and is now facing t-AML with del(17) on karyotype and
TP53 mutation on next-generation sequencing (NGS) should be screened for Li–Fraumeni syndrome (even if her family history is not indicative of it) [
22]. Knowing that an important subgroup of younger patients with t-AML carry germline mutations in cancer predisposition genes is important not only for their treatment (e.g., selection of appropriate family donors for allogeneic HCT) but also for identifying family members who may be at high risk for the development of tumors.
Studies have also examined whether polymorphisms in genes involved in the metabolism of alkylating agents and topoisomerase II inhibitors such as cytochrome P450 enzymes (CYP3A4/CYP3A5), GSTM1, GSTT1, and NQO1 may confer an increased risk of t-AML, with so far controversial results [
10,
27].
3.2. Current Model for the Molecular Pathogenesis of t-AML
Beyond genetic predisposition and the inheritance of familial cancer genes, NGS has revealed that, in many patients, the first step in the process of t-AML development is age-related clonal hematopoiesis also known as clonal hematopoiesis of indeterminate potential (CHIP) [
28]. According to this model, HSC clones harboring somatic
TP53 or
PPM1D mutations are detected in patients before chemotherapy exposure [
29,
30]. Owing to its competitive advantage, the
TP53-mutant clone enlarges in the bone marrow after chemotherapy administered for a neoplastic or non-neoplastic disorder. The acquisition of additional mutations or the emergence of cytogenetic abnormalities such as chromosome 5/5q or 7/7q deletion leads to a selection of subclones of hematopoietic cells with an increasingly impaired differentiation capacity, which drives leukemic transformation (
Figure 1) [
8,
22,
31].
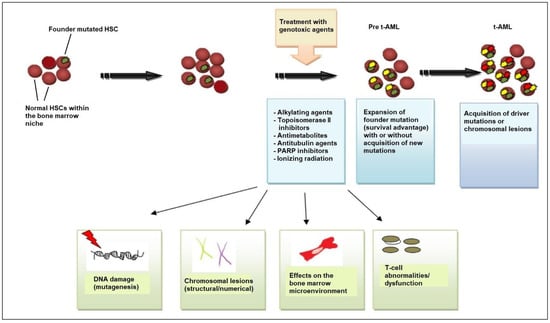
Figure 1. Pathogenesis of t-AML. Evidence supports the view that chemotherapy and/or radiation selects for the expansion of a mutant clone (founder clone) in the hematopoietic-stem-cell (HSC) and/or progenitor-cell compartment of the bone marrow that is more resistant to DNA damage. After chemotherapy/radiotherapy exposure, the pool of normal HSCs is affected and depleted, allowing the mutant clone to expand, owing to its survival advantage. Cytotoxic therapy affects the stem cell, causing DNA damage (i.e., additional mutations and/or chromosomal abnormalities) that drives the emergence of fully leukemic clones and changes in the bone marrow microenvironment, including bone marrow stromal cells and T-cell subsets (cytotoxic T-cells, regulatory T-cells). Stromal cells support HSC function but, if challenged with genotoxic agents, can inhibit normal HSC survival, providing an opportunity for the selection of pre-leukemic clones, and T-cell dysfunction interferes with tumor immunosurveillance. All these mechanisms contribute to the development of leukemic cells and ultimately to the emergence of t-AML. Abbreviations: t-AML, therapy-related acute myeloid leukemia; PARP, poly (ADP-ribose) polymerase.
The mutational burden in t-AML is similar to de novo AML, but the relative frequency of specific mutations differs significantly, e.g., mutations in the gene encoding nucleophosmin (
NPM1) are not as common in t-AML as in de novo AML [
16,
32,
33]. Notably,
TP53 mutations are the most common molecular abnormality in t-AML.
3.3. TP53 Mutations
The
TP53 tumor-suppressor gene, located on the short arm of chromosome 17, encodes a 53-kd nuclear phosphoprotein that suppresses cell growth in response to DNA damage through several mechanisms including cell-cycle arrest at the G1/S checkpoint, activation of DNA-repair enzymes, and initiation of apoptosis in cases of severe DNA damage [
9]. From a mechanistic point of view, the activation of the DNA damage checkpoint results in the formation of
TP53 homo-tetramers (tetramerization is essential for p53 activation in vivo). Activated
TP53 then interacts with other tumor suppressors such as p21
Waf1/Cip1, p63, and p73, activates numerous microRNAs (including the miR-34 family of miRNAs) and proapoptotic proteins (BCL-XL, BCL2, Bax), and alters the function of mTOR kinase.
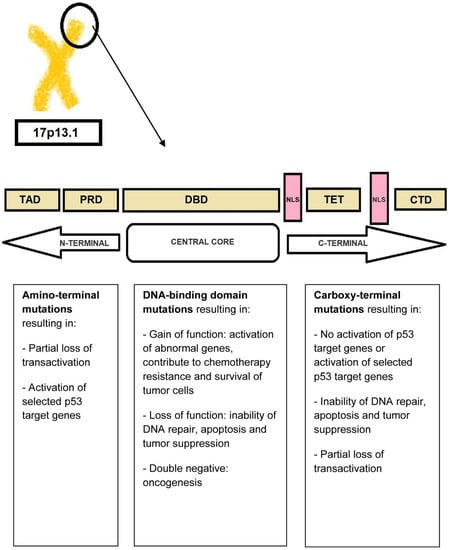
The transcriptional activity of
TP53 is carried out by five distinct domains including the transactivation activation domain (TAD) and the proline-rich domain (PRD), located at the N-terminal portion of the p53 protein, the core DNA-binding domain (DBD) and the tetramerization domain (TET) (responsible for the oligomerization of the p53 protein, which exists as a tetramer), and the carboxy-terminal regulatory domain (CTD) at the C-terminal portion of the protein. The C-terminal portion also includes several nuclear localization sequences (NLS) (
Figure 2) [
34,
35].
Figure 2. P53 protein structure. The N-terminal portion of the p53 protein includes the transactivation domains (TAD) I and II and the proline-rich domain (PRD). The central core of the protein includes the DNA-binding domain (DBD). The C-terminal portion of p53 includes the tetramerization domain (TET) required for p53 tetramerization and activation, several nuclear localization sequences (NLS), and the carboxy-terminal regulatory domain.
TP53 is mutated in a large proportion of tumors [
34,
36]. In fact, acquired
TP53 mutations represent the most common specific genetic change in human cancer. The types of mutations affecting
TP53 include gain-of-function, loss-of-function, and separation-of-function mutations [
37,
38]. Approximately 18,000 different mutations have been found in different types of malignancies. It is noteworthy that
TP53 shows a specific set of mutations depending on the type of malignancy [
34,
39]. Mutations that deactivate p53 (loss-of-function mutations) usually occur in the DBD. Most of these mutations impair the ability of the protein to bind to its target DNA sequences, thus preventing the transcriptional activation of p53 target genes. The loss of
TP53 function confers a clonal advantage [
9].
In t-AML, the usual mutation (>80%) is a monoallelic missense mutation (i.e., a point mutation in which a single nucleotide is substituted by another, leading to the replacement of a single amino acid) in the DNA-binding domain [
40,
41,
42]. The result of this mutation is the abnormally increased expression of the mutant p53 protein due to a longer half-life compared to the wild-type protein (in normal tissues, p53 protein is present in very low quantities so that it is not readily detectable by immunochemistry. However, in
TP53-mutated leukemic cells, large amounts of p53 protein accumulate in the nucleus which can be seen by staining [“p53 over-expression”]; this is generally attributed to the accumulation of over-stabilized, mutant protein). Less frequent mutations include deletions, truncations, insertions, nonsense, and splice-site mutations [
40,
41,
43]. The definition of t-AML with
TP53 mutation requires the presence of a somatic
TP53 mutation with a variant-allele frequency [VAF] of >10%. Monoallelic
TP53-mutated t-AML has a poor prognosis. The term “multi-hit” mutations refers to the presence of two distinct
TP53 mutations (each with a VAF of >10%) or a single
TP53 mutation with either (i) 17/17p deletion on cytogenetics; (ii) copy-neutral loss of heterozygosity (LOH) at the 17p (
TP53 locus); or (iii) VAF of >50%, which is 75% concordant with copy-neutral LOH variants [
44,
45].
Different
TP53 mutations occur at different stages in the course of AML. For example, biallelic defects and multi-hit events take place at a very early stage during leukemogenesis. These mutations contribute to the creation of a dominant clone [
46,
47]. Monoallelic
TP53 mutations, on the other hand, usually occur at later stages of the disease as subclonal events [
46,
47]. Monoallelic
TP53 mutations may also coexist with other driver mutations such as
SF3B1,
TET2,
DMT3A, and
ASXL1 mutations [
46,
47].
TP53 mutations are associated with complex or monosomal karyotypes and “chromosome shattering” (also known as “chromothripsis”), a phenomenon characterized by extensive chromosomal rearrangements [
48,
49,
50].
An important development is the discovery that p53 activates the transcription of a set of microRNAs, including the miR-34 family. Pathogenic mutations in
TP53 result in diminished expression of microRNA-34a (miR-34a), a potent tumor-suppressive microRNA, leading to the over-expression of
c-MYC oncogene and upregulation of PD-L1 in tumor cells [
22,
51].
3.4. Molecular Basis of t-AML
Lindsley and coworkers investigated the genetic basis of t-AML and secondary AML (s-AML) and identified three distinct subtypes of somatic mutations: (i) secondary-type mutations involving eight genes (
SRSF2,
SF3B1,
U2AF1,
ZRSR2,
ASXL1,
EZH2,
BCOR, and
STAG2) which are commonly linked to MDS and are now recognized as “AML with myelodysplasia-related (MR) gene mutation” in 2022 ELN recommendations; (ii)
TP53 mutations, which are associated with complex karyotypes (often monosomal, with frequent abnormalities of chromosomes 5 and 7), intrinsic therapy resistance, and very poor survival; and (iii) “de novo”-type (or pan-AML-type) mutations including
NPM1 mutations,
KMT2A (MLL) rearrangements at 11q23 locus, core-binding-factor (CBF) chromosomal rearrangements, myeloid-transcription-factor mutations (e.g.,
RUNX1,
CEBPA, and
GATA2), signal-transduction-protein mutations (e.g.,
FLT3,
N-RAS, and
K-RAS), and other mutations (e.g.,
IDH1, IDH2, and
WT1) [
8].
“Secondary-type” mutations are found in 30% of t-AML cases and are associated with poor outcomes.
TP53 mutations are seen in ~50% of t-AML cases.
NPM1 mutations are identified in 5.4% of patients without concurrent “secondary-type” or
TP53 mutations [
8,
30,
52], suggesting that there is a non-random pattern of co-mutations with mutual exclusivity between
TP53 and
NPM1 mutations in t-AML. Patients with t-AML with “secondary-type” mutations are significantly older than patients with “de novo”/pan-AML mutations. Clinically, t-AML with “secondary-type” mutations closely resembles s-AML. In contrast, patients with t-AML with “de novo”-type mutations closely resemble patients with primary de novo AML [
8,
53].
Recently, Papaemmanuil and coworkers presented another categorization of patients with AML into 16 distinct molecular subgroups [
54]. Regarding t-AML, the majority of patients had high relapse rates and poor prognosis, regardless of the achievement of early minimal residual disease (MRD) negativity. However, beyond this general rule, patients with t-AML who achieved CR, including those with
TP53 mutations, seemed to benefit from HCT. Additionally, patients who had >2 mutations had worse prognosis compared to those who carried a single-gene mutation. The prognosis was even worse in patients with
TP53 mutations or inv(3)/t(3;3), resulting in deregulated
MECOM (
EVI1) and
GATA2 expression [
54]. However, it should be emphasized that even patients carrying
TP53 mutations benefited from HCT, especially in CR1 (and less so after CR2) [
54].
Clearly, the past decade has reshaped our view of t-AML. Rather than considering t-AML as one clinical entity, it is more appropriate to view t-AML as at least three molecular types (according to the Lindsley model) that vary in prevalence with age, each bearing more similarity to AML with the same genetic alterations and no leukemogenic exposure.
3.5. A Permissive Bone Marrow Microenvironment Facilitates t-AML Growth
The immune system inside the bone marrow is also involved in the development of t-AML. Population-based studies show that AML is more common among patients with autoimmune diseases than the general population [
55,
56]. The risk of developing t-AML appears to be related to the type of autoimmune disease and type and duration of its treatment. Drugs used to treat autoimmune diseases such as azathioprine, mitoxantrone, and cyclophosphamide may directly damage DNA and increase the risk of leukemogenesis [
55,
56]. However, drugs used in autoimmune disorders such as azathioprine may also affect the balance between T-cell subsets. Experiments in mice have shown that high doses of azathioprine downregulate regulatory T-cells (T-regs), whereas lower doses upregulate T-regs [
57]. Abnormalities in T-regs may play a role in the transition of MDS to AML [
58,
59]. For example, in patients with clinically stable MDS (regardless of the disease stage), T-reg levels remain stable, but upon transformation to AML, an increase in T-reg numbers is noted both in the marrow and in the peripheral blood [
60]. In addition, reduced immune surveillance by cytotoxic T-cells is seen in patients with autoimmune disorders. Chronic inflammatory signaling and inflammatory conditions can modulate the bone marrow microenvironment and facilitate the survival and proliferation of leukemic cells [
56,
61]. Notably, the transcription factor nuclear factor-κB (NF-κB), a central pro-inflammatory mediator, and polymorphisms in the interleukin-1 receptor antagonist (IL-1Rα) are involved in both autoimmune diseases and leukemogenesis [
56,
62,
63,
64,
65].
3.6. Role of Pro-Inflammatory Cytokine Signaling
The response of a body to a cancer is not a unique mechanism but has many parallels with the chronic inflammation seen in chronic infections. Balkwill and Mantovani’s metaphor, stating that if genetic damage is “the match that lights the fire of cancer”, some types of inflammation provide “the fuel that feeds the flames”, puts emphasis on inflammation as a major contributor in the growth of some cancers [
66]. Multifaceted activation of the immune system and chronic inflammation accompany many hematologic neoplasms including MDS and AML. Certain pro-inflammatory cytokines such as the tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6) play an important role in the development of AML. IL-1 occurs as two structurally related polypeptides (IL-1α and IL-1β), each of which has a broad spectrum of both beneficial and harmful biologic effects. IL-1β, in particular, contributes to the proliferation and survival of leukemic cells [
65,
67]. It has been found that IL-1β participates in the immune response through a dual mechanism: the activation of the IL-1 receptor (IL-1R)/Toll-like receptor (TLR) and caspase-1 activation [
68,
69]. The activation of IL-1β induces a signaling cascade that leads to the phosphorylation and ubiquitination of MyD88, IRAK-4, and TRAF-6 and, ultimately, NF-κB activation [
70]. At the same time, IL-1β induces the activation of p38 mitogen-activated protein kinase (MAPK), as well as the activation of the transcription factor
GATA2 [
70,
71,
72,
73]. Both p38 MAPK and
GATA2 contribute to the proliferation of leukemic blasts. Monocytes and macrophages represent the main cell source of IL-1β. IL-1β may be a target for future therapies in AML.
IL-35, transforming growth factor β (TGF-β), and IL-10 have also been implicated in the pathogenesis of AML. IL-35 belongs to the IL-12 family and is produced by T-regs. In patients with AML, high concentrations of IL-35 have been found in bone marrow plasma, corresponding to an increased proportion of T-regs [
74,
75,
76]. TGF-β contributes to many cellular processes including cell survival, proliferation, and migration. In AML, TGF-β inhibits the proliferation of leukemic stem cells (LSCs), maintaining their longevity.
The administration of DNA damaging agents, in association with disturbances in T-cell subsets and pro-inflammatory changes in the bone marrow microenvironment, may favor the development of t-AML in patients with autoimmune disorders.