Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rainer Schulz | -- | 1718 | 2023-04-06 11:50:23 | | | |
| 2 | Catherine Yang | Meta information modification | 1718 | 2023-04-07 03:04:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Schulz, R.; Schlüter, K. Monoamine Oxidases. Encyclopedia. Available online: https://encyclopedia.pub/entry/42839 (accessed on 24 July 2026).
Schulz R, Schlüter K. Monoamine Oxidases. Encyclopedia. Available at: https://encyclopedia.pub/entry/42839. Accessed July 24, 2026.
Schulz, Rainer, Klaus-Dieter Schlüter. "Monoamine Oxidases" Encyclopedia, https://encyclopedia.pub/entry/42839 (accessed July 24, 2026).
Schulz, R., & Schlüter, K. (2023, April 06). Monoamine Oxidases. In Encyclopedia. https://encyclopedia.pub/entry/42839
Schulz, Rainer and Klaus-Dieter Schlüter. "Monoamine Oxidases." Encyclopedia. Web. 06 April, 2023.
Copy Citation
On the one hand, reactive oxygen species (ROS) are involved in the onset and progression of a wide array of diseases. On the other hand, these are a part of signaling pathways related to cell metabolism, growth and survival. While ROS are produced at various cellular sites, in cardiomyocytes the largest amount of ROS is generated by mitochondria. Apart from the electron transport chain and various other proteins, monoamine oxidases (MAO) has been proposed to modify mitochondrial ROS formation.
ischemia
reperfusion
heart failure
pulmonary hypertension
1. Monoamine Oxidase Isoforms
Two different isoforms of MAO are known, namely MAO-A and MAO-B, both of which are located at the outer mitochondrial membrane. Species-dependent cardiomyocytes express different MAO isoforms: in rats, MAO-A predominates in adulthood, [1][2] while in adult mice MAO-B dominates [3][4]. Interestingly, in rat hearts, MAO-B activity also predominates up to an age of 2–3 weeks [5], most likely since MAO-B expression increases under mechanical strain as compared to the quiescent situation [6]. Human cardiomyocytes contain both MAO isoforms, but with more, albeit moderate, expression for MAO-A [7][8]. In rat hearts, MAO activity is higher in the left compared to the right ventricle [9] and females have higher plasma MAO activity than males [10] as estrogens can modulate MAO activity [11].
2. MAO Substrates
The two MAO isoforms have common substrates such as dopamine, but also specific substrates: MAO-B can metabolize 1-methyl histamine [12], produced by the histamine-N-methyltransferase [13], while MAO-A metabolizes serotonin (or 5-hydroxytryptamin, 5-HT) and catecholamines (for review, see [14]). MAO requires flavin adenine dinucleotide as a cofactor that is reduced by the reaction of, and subsequently re-oxidized by, oxygen and water, generating hydrogen peroxide [15]. MAO can also form reactive aldehydes, such as 4-hydroxynonenal, as a byproduct of catecholamine metabolism through cardiolipin peroxidation inside mitochondria in primary cardiomyocytes. Deleterious effects of 4-hydroxynonenal are physiologically prevented by the activation of mitochondrial aldehyde dehydrogenase 2 [16].
Mice deficient in both MAO-A and MAO-B demonstrate increased tissue levels of serotonin, norepinephrine, dopamine, and phenylethylamine [17], and genetic ablation of MAO-A increases the serotonin concentration in the blood and tissue of rats [18]. Similarly, a blockade of MAO by drugs indicated for other uses (e.g., antidepressants) can alter histamine levels in mice hearts [19]. In contrast, MAO-A overexpression decreases the level of norepinephrine and serotonin in the heart (for review, see [20]) (Figure 1).
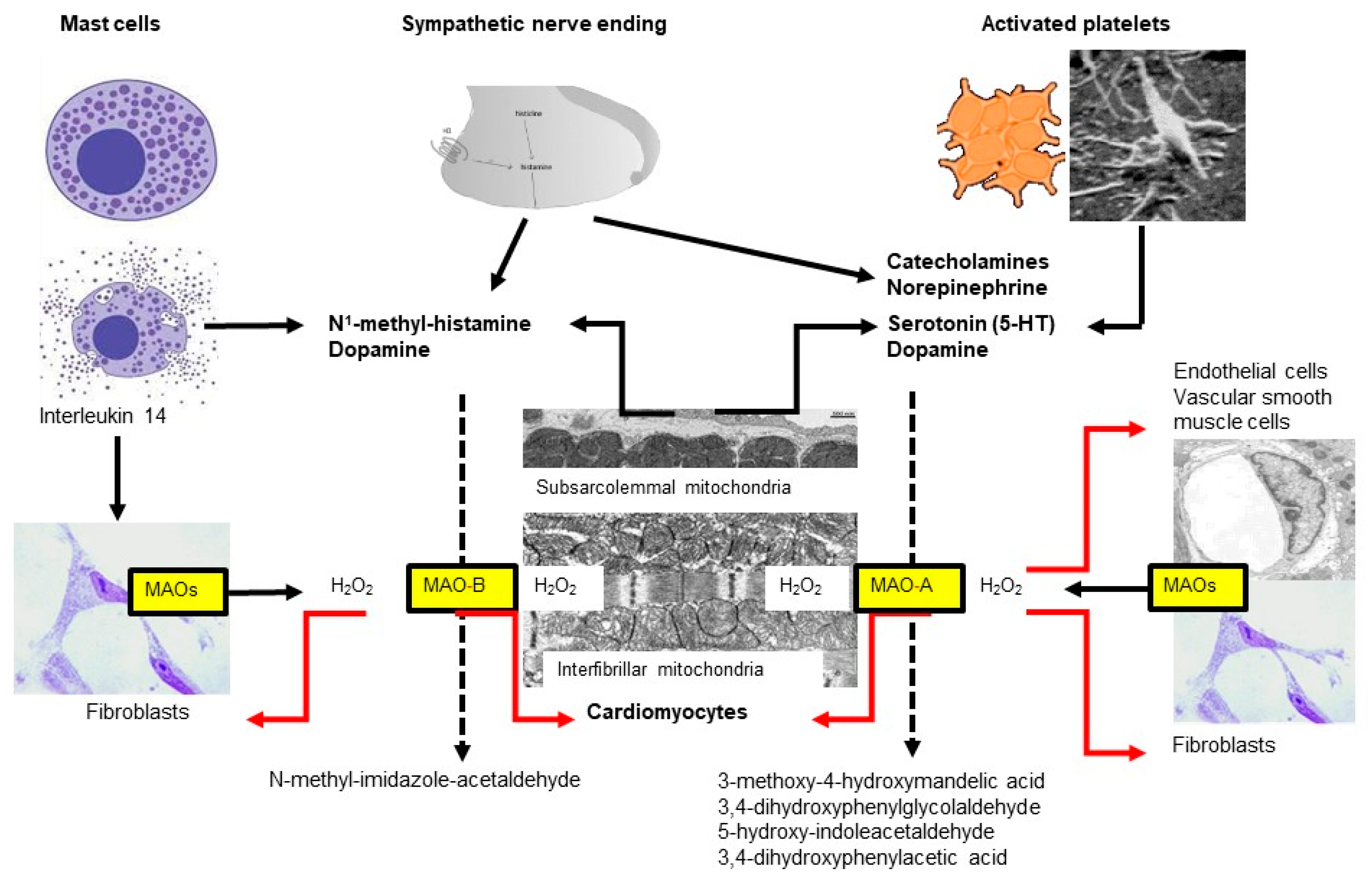
Figure 1. Two subtypes of monoamine oxidases (MAO)—named A and B—are located at the outer mitochondrial membrane, which differ in their substrate specificity. Almost all cell types express MAOs but the respective subtype might differ between species, organs and age. In the heart, MAOs are expressed in cardiomyocytes, fibroblasts, vascular smooth muscle and endothelial cells. In the heart, MAO substrates are derived from different sources including mast cells, sympathetic nerves, platelets and cardiomyocytes.
3. MAO Expression
An increased expression/activity of MAO occurs during aging [21][22] and with different diseases such as arterial hypertension [23][24], pulmonary hypertension [25][26], hypertrophy [27], diabetes [28][29][30][31], myocardial infarction [32] or heart failure [33][34]. In the streptozotocin-induced diabetic rat model, particularly the MAO-B isoform is induced in aortas and hearts and contributes to the generation of reactive oxygen species [28]. While the underlying mechanisms of MAO upregulation under the above conditions are still unclear, one potential factor contributing to increased MAO expression/activity in the heart might be increased substrate availability (for review, see [20]).
An increased sympathetic tone increases plasma norepinephrine and epinephrine concentrations, and increased norepinephrine spillover as seen in chronic heart failure patients [35][36]. Serotonin concentrations are increased during different disease states (for review, see [37]) and part of the increase has been attributed to altered platelet function [38]. Histamine co-localizes with norepinephrine in neurons [39] and is enclosed in cytoplasmatic granules of mast cells, which lie adjacent to blood vessels and between cardiomyocytes [40], and mast cell degranulation might occur under stress conditions [41]. Moreover, serotonin can be formed in the mouse and human heart [42], probably by cardiomyocytes themselves [43].
In AC16 cardiomyocytes, MAO-A mRNA and protein expressions are affected by non-coding RNAs since knockdown of the non-protein coding RNA 472 (LINC00472) reduced MAO-A expression, the results being partly abolished by miR-335-3p inhibition. Thus, LINC00472 positively regulates MAOA expression via interaction with miR-335-3p [44].
4. Monoamine Oxidases and Hypertrophy
Cardiac hypertrophy is a typical early adaptive response to increased cardiac workload and mechanical stress. However, in cases of prolonged or chronic stress, this response may become maladaptive and ultimately lead to heart failure. Cardiomyocytes synthesize additional sarcomeres, leading to the thickening of the ventricular wall and increased overall cardiac mass and size. The subcellular reorganization that underlies cardiomyocyte hypertrophy was found to require functional and responsive mitochondrial dynamics (for review, see [45]).
In wild type mice, pressure overload induced by transverse aortic constriction results in increased dopamine catabolism, left ventricular hypertrophy and dilation progressing to cardiac dysfunction. In contrast, in MAO-B knockout mice with transverse aortic constriction concentric left ventricular hypertrophy and function are maintained, both at the early (weeks) and late stages (months) [46]. As outlined above, in the hearts [23] as well as in the isolated cardiomyocytes [24] of spontaneously hypertensive rats MAO activity is significantly increased even before the development of cardiac hypertrophy [24]. Increased MAO activity might represent an early event in the development of cardiac hypertrophy [23] due to its potential impact on cardiac metabolism [47] since cardiac hypertrophy normally goes along with a metabolic switch to preferential use of carbohydrates rather than fatty acids [48][49].
In rat cardiomyocytes, administration of high micromolar concentrations of serotonin or dopamine increases glucose transport through the upregulation of glucose transporters 1 and 4 at the sarcolemma; the increase in glucose import is blocked by MAO-A inhibition [47]. At similar concentrations, serotonin induces cardiomyocyte hypertrophy, again the effect being largely attenuated by MAO-A inhibition [50] (or blockade of the extracellular regulated kinase). In addition, the effects of angiotensin II on hypertrophy are attenuated by a pharmacological blockade of MAO-A in rats [51]. Since lower concentrations of serotonin induce cardiomyocyte hypertrophy independent of MAO-A through the activation of the 5-HT(A2) receptor [52][53], genetic deletion of MAO-A also increases load-dependent ventricular hypertrophy [18] (for review, see [2]). Thus, both an increased or decreased MAO expression/activity can contribute to hypertrophic effects, depending on substrate availability.
5. Pulmonary Hypertension
MAOs have also been proposed to play an important role in pulmonary hypertension [54]. In rats, pulmonary hypertension secondary to monocrotaline injection [25], sugen5416/hypoxia, or pulmonary artery banding [26] upregulates MAO-A expression in the pulmonary vasculature and the failing right ventricle. Clorgyline treatment reduced the right ventricular afterload and pulmonary vascular remodeling in sugen/hypoxia rats through reduced pulmonary vascular proliferation and oxidative stress, resulting in improved right ventricular stiffness and relaxation and reversed right ventricular hypertrophy. In rats with pulmonary artery banding, clorgyline had no direct effect on the right ventricle [26]. In contrast, recent unpublished data demonstrate less myocardial structural or functional changes secondary to the pulmonary artery banding in cardiomyocyte-specific MAO B knockout mice in the right ventricle.
6. Monoamine Oxidases and Ischemia/Reperfusion (I/R) Injury
Under stress conditions such as I/R, the autonomic nervous system is activated, releasing neurotransmitters that are metabolized by MAOs, thereby directly influencing heart function [55]. Besides norepinephrine, serotonin and histamine also play important roles in I/R injury. Serotonin accumulates in the heart during ischemia [56] and is degraded after reperfusion depending on MAO-A activity after uptake into cells [57]. Mast cells become activated during stress conditions and release histamine [41]; histamine release from the heart is increased during I/R [12]. While mast cell activation thus increases substrate availability for MAO-B, MAO-B inhibition prevents mast cell degranulation in diabetic mice hearts [30], implying a vicious cycle of mast cell and MAO-B activation.
During 30 min ischemia, hydroxyl radical production increases 2-fold with a further increase upon 60 min reperfusion in isolated rat hearts, both of which can be decreased by pargyline administration. The decrease in ROS formation following MAO inhibition is associated with reduced cardiomyocyte injury following I/R [58]. Similarly, cardiomyocyte-specific MAO-B knockout reduces infarct size following I/R in isolated mice hearts [59].
In vivo, the inhibition of MAO-A largely reduces myocardial ultrastructural damage induced by 30 min ischemia and 60 min reperfusion in the rat heart, associated with the prevention of postischemic oxidative stress, neutrophil accumulation, and mitochondrial-dependent cell death [60]. Infarct size and cardiomyocyte apoptosis are also significantly decreased in MAO-A-deficient animals following 30 min ischemia and 180 min reperfusion, the protection being accompanied by sphingosine kinase 1 inhibition and less ceramide accumulation [61].
Cardioprotection can be achieved also by mechanical intervention such as ischemic preconditioning [62]. In both male and female rat hearts, ischemic preconditioning improves functional recovery following I/R, which is further enhanced in the presence of MAO inhibition by either clorgyline or pargyline. However, infarct size is similar among all preconditioned groups, regardless of the presence of MAO inhibitors, indicating that acute inhibition of MAOs potentiates the preconditioning-induced postischemic functional recovery without having any further effect on infarct size beyond that achieved by ischemic preconditioning [63].
7. Monoamine Oxidases and Left Ventricular Remodeling/Heart Failure
Rasagiline mesylate (N-propargyl-1 (R)-aminoindan) (RG), a selective, potent irreversible inhibitor of monoamine oxidase-B, administered for 28 days (2 mg/kg) starting 24 h after myocardial infarction, preserves left ventricular geometry and function. Treatment with rasagiline prevents tissue fibrosis and attenuates cardiomyocyte apoptosis in the border zone of the infarct associated with a markedly-decreased malondialdehyde level in the border zone, indicating a reduction in tissue oxidative stress [64]. Additionally, MAO-A is an important source of oxidative stress in the heart and MAO-A-derived reactive oxygen species contribute to dilated cardiomyopathy [31]. In mice, left ventricular function following four weeks of coronary artery occlusion improves by pharmacological or genetic inhibition of MAO-A. Both interventions protect the mice from 4-hydroxynonenal accumulation and mitochondrial calcium overload, thus mitigating ventricular dysfunction [16].
Furthermore, it has recently been suggested that upregulation of MAO-A during heart failure will accelerate intracellular catecholamine degradation, thereby inhibiting a direct stimulation of β-adrenergic receptors at the sarcoplasmic reticulum; this interaction is closely linked to phospholamban phosphorylation and calcium filling of the sarcoplasmic reticulum [65][66].
The importance of MAO for left ventricular remodeling and heart failure development can be shown in mice with chronic overexpression of MAO-A. Here, reactive oxygen species [22] and 4-hydroxynonenal concentrations increase, followed by mitochondrial dysfunction [67], cardiomyocyte hypertrophy, reduced left ventricular function and increased cardiac fibrosis [16], as well as increased cardiac inflammation [68] (for review, see [20][69]). When transgenic animals are treated with the antioxidant N-acetyl cysteine part of the above effects can be rescued [22][70].
Moreover, heart failure induced by doxorubicin is affected by MAO inhibition, which prevents both the severe oxidative stress induced by doxorubicin as well as chamber dilation and cardiac dysfunction in doxorubicin-treated mice in vivo [71].
References
- Edwards, D.J.; Koon Yan, P.; Venetti, M.C. Developmental aspects of rat heart monoamine oxidase. Biochem. Pharmacol. 1979, 28, 2337–2343.
- Monassier, L.; Laplante, M.A.; Ayadi, T.; Doly, S.; Maroteaux, L. Contribution of gene-modified mice and rats to our understanding of the cardiovascular pharmacology of serotonin. Pharmacol. Ther. 2010, 128, 559–567.
- Saura, J.; Richards, J.G.; Mahy, N. Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Neurobiol. Aging 1994, 15, 399–408.
- Strolin Benedetti, M.; Thomassin, J.; Tocchetti, P.; Dostert, P.; Kettler, R.; Da Prada, M. Species differences in changes of heart monoamine oxidase activities with age. J. Neural. Transm. Suppl. 1994, 41, 83–87.
- Strolin Benedetti, M.; Dostert, P.; Tipton, K.F. Developmental aspects of the monoamine-degrading enzyme monoamine oxidase. Dev. Pharmacol. Ther. 1992, 18, 191–200.
- Ohki, R.; Yamamoto, K.; Ueno, S.; Mano, H.; Ikeda, U.; Shimada, K. Effects of olmesartan, an angiotensin II receptor blocker, on mechanically-modulated genes in cardiac myocytes. Cardiovasc. Drugs Ther. 2003, 17, 231–236.
- Rodriguez, M.J.; Saura, J.; Billett, E.E.; Finch, C.C.; Mahy, N. Cellular localization of monoamine oxidase A and B in human tissues outside of the central nervous system. Cell Tissue Res. 2001, 304, 215–220.
- Sivasubramaniam, S.D.; Finch, C.C.; Rodriguez, M.J.; Mahy, N.; Billett, E.E. A comparative study of the expression of monoamine oxidase-A and -B mRNA and protein in non-CNS human tissues. Cell Tissue Res. 2003, 313, 291–300.
- Meco, M.; Bonifati, V.; Collier, W.L.; Ramacci, M.T.; Amenta, F. Enzyme histochemistry of monoamine oxidase in the heart of aged rats. Mech. Ageing Dev. 1987, 38, 145–155.
- Robinson, D.S.; Davis, J.M.; Nies, A.; Ravaris, C.L.; Sylwester, D. Relation of sex and aging to monoamine oxidase activity of human brain, plasma, and platelets. Arch. Gen. Psychiatry 1971, 24, 536–539.
- Zhang, Z.; Chen, K.; Shih, J.C.; Teng, C.T. Estrogen-related receptors-stimulated monoamine oxidase B promoter activity is down-regulated by estrogen receptors. Mol. Endocrinol. 2006, 20, 1547–1561.
- Costiniti, V.; Spera, I.; Menabo, R.; Palmieri, E.M.; Menga, A.; Scarcia, P.; Porcelli, V.; Gissi, R.; Castegna, A.; Canton, M. Monoamine oxidase-dependent histamine catabolism accounts for post-ischemic cardiac redox imbalance and injury. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864 Pt B, 3050–3059.
- Maintz, L.; Novak, N. Histamine and histamine intolerance. Am. J. Clin. Nutr. 2007, 85, 1185–1196.
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural. Transm. (Vienna) 2018, 125, 1627–1634.
- Mialet-Perez, J.; Bianchi, P.; Kunduzova, O.; Parini, A. New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J. Neural. Transm. (Vienna) 2007, 114, 823–827.
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yucel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923.
- Holschneider, D.P.; Scremin, O.U.; Roos, K.P.; Chialvo, D.R.; Chen, K.; Shih, J.C. Increased baroreceptor response in mice deficient in monoamine oxidase A and B. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H964–H972.
- Lairez, O.; Calise, D.; Bianchi, P.; Ordener, C.; Spreux-Varoquaux, O.; Guilbeau-Frugier, C.; Escourrou, G.; Seif, I.; Roncalli, J.; Pizzinat, N.; et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J. Mol. Cell. Cardiol. 2009, 46, 587–595.
- Neumann, J.; Grobe, J.M.; Weisgut, J.; Schwelberger, H.G.; Fogel, W.A.; Marusakova, M.; Wache, H.; Bahre, H.; Buchwalow, I.B.; Dhein, S.; et al. Histamine can be Formed and Degraded in the Human and Mouse Heart. Front. Pharmacol. 2021, 12, 582916.
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42.
- Maurel, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Frances, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1460–H1467.
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox Signal. 2013, 18, 5–18.
- Tanijiri, H. Cardiac hypertrophy in spontaneously hypertensive rats. Jpn. Heart J. 1975, 16, 174–188.
- Pino, R.; Failli, P.; Mazzetti, L.; Buffoni, F. Monoamine oxidase and semicarbazide-sensitive amine oxidase activities in isolated cardiomyocytes of spontaneously hypertensive rats. Biochem. Mol. Med. 1997, 62, 188–196.
- van Eif, V.W.; Bogaards, S.J.; van der Laarse, W.J. Intrinsic cardiac adrenergic (ICA) cell density and MAO-A activity in failing rat hearts. J. Muscle Res. Cell Motil. 2014, 35, 47–53.
- Sun, X.Q.; Peters, E.L.; Schalij, I.; Axelsen, J.B.; Andersen, S.; Kurakula, K.; Gomez-Puerto, M.C.; Szulcek, R.; Pan, X.; da Silva Goncalves Bos, D.; et al. Increased MAO-A Activity Promotes Progression of Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 331–343.
- Strom, C.C.; Kruhoffer, M.; Knudsen, S.; Stensgaard-Hansen, F.; Jonassen, T.E.; Orntoft, T.F.; Haunso, S.; Sheikh, S.P. Identification of a core set of genes that signifies pathways underlying cardiac hypertrophy. Comp. Funct. Genom. 2004, 5, 459–470.
- Sturza, A.; Duicu, O.M.; Vaduva, A.; Danila, M.D.; Noveanu, L.; Varro, A.; Muntean, D.M. Monoamine oxidases are novel sources of cardiovascular oxidative stress in experimental diabetes. Can. J. Physiol. Pharmacol. 2015, 93, 555–561.
- Manni, M.E.; Zazzeri, M.; Musilli, C.; Bigagli, E.; Lodovici, M.; Raimondi, L. Exposure of cardiomyocytes to angiotensin II induces over-activation of monoamine oxidase type A: Implications in heart failure. Eur. J. Pharmacol. 2013, 718, 271–276.
- Deshwal, S.; Forkink, M.; Hu, C.H.; Buonincontri, G.; Antonucci, S.; Di Sante, M.; Murphy, M.P.; Paolocci, N.; Mochly-Rosen, D.; Krieg, T.; et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. 2018, 25, 1671–1685.
- Umbarkar, P.; Singh, S.; Arkat, S.; Bodhankar, S.L.; Lohidasan, S.; Sitasawad, S.L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic. Biol. Med. 2015, 87, 263–273.
- Jin, H.; Yang, R.; Awad, T.A.; Wang, F.; Li, W.; Williams, S.P.; Ogasawara, A.; Shimada, B.; Williams, P.M.; de Feo, G.; et al. Effects of early angiotensin-converting enzyme inhibition on cardiac gene expression after acute myocardial infarction. Circulation 2001, 103, 736–742.
- Kong, S.W.; Bodyak, N.; Yue, P.; Liu, Z.; Brown, J.; Izumo, S.; Kang, P.M. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol. Genom. 2005, 21, 34–42.
- Petrak, J.; Pospisilova, J.; Sedinova, M.; Jedelsky, P.; Lorkova, L.; Vit, O.; Kolar, M.; Strnad, H.; Benes, J.; Sedmera, D.; et al. Proteomic and transcriptomic analysis of heart failure due to volume overload in a rat aorto-caval fistula model provides support for new potential therapeutic targets—monoamine oxidase A and transglutaminase 2. Proteome Sci. 2011, 9, 69.
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762.
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753.
- Rieder, M.; Gauchel, N.; Bode, C.; Duerschmied, D. Serotonin: A platelet hormone modulating cardiovascular disease. J. Thromb. Thrombolysis 2021, 52, 42–47.
- Nigmatullina, R.R.; Kirillova, V.V.; Jourjikiya, R.K.; Mukhamedyarov, M.A.; Kudrin, V.S.; Klodt, P.M.; Palotas, A. Disrupted serotonergic and sympathoadrenal systems in patients with chronic heart failure may serve as new therapeutic targets and novel biomarkers to assess severity, progression and response to treatment. Cardiology 2009, 113, 277–286.
- He, G.; Hu, J.; Li, T.; Ma, X.; Meng, J.; Jia, M.; Lu, J.; Ohtsu, H.; Chen, Z.; Luo, X. Arrhythmogenic effect of sympathetic histamine in mouse hearts subjected to acute ischemia. Mol. Med. 2012, 18, 1–9.
- Genovese, A.; Spadaro, G. Highlights in cardiovascular effects of histamine and H1-receptor antagonists. Allergy 1997, 52 (Suppl. S34), 67–78.
- He, Z.; Ma, C.; Yu, T.; Song, J.; Leng, J.; Gu, X.; Li, J. Activation mechanisms and multifaceted effects of mast cells in ischemia reperfusion injury. Exp. Cell Res. 2019, 376, 227–235.
- Gergs, U.; Jung, F.; Buchwalow, I.B.; Hofmann, B.; Simm, A.; Treede, H.; Neumann, J. Pharmacological and physiological assessment of serotonin formation and degradation in isolated preparations from mouse and human hearts. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1087–H1097.
- Ponicke, K.; Gergs, U.; Buchwalow, I.B.; Hauptmann, S.; Neumann, J. On the presence of serotonin in mammalian cardiomyocytes. Mol. Cell. Biochem. 2012, 365, 301–312.
- Mo, G.; Mo, J.; Tan, X.; Wang, J.; Yan, Z.; Liu, Y. Yin Yang 1 (YY1)-induced long intergenic non-protein coding RNA 472 (LINC00472) aggravates sepsis-associated cardiac dysfunction via the micro-RNA-335-3p (miR-335-3p)/Monoamine oxidase A (MAOA) cascade. Bioengineered 2022, 13, 1049–1061.
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076.
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid. Redox Signal. 2014, 20, 267–280.
- Fischer, Y.; Thomas, J.; Kamp, J.; Jungling, E.; Rose, H.; Carpéné, C.; Kammermeier, H. 5-hydroxytryptamine stimulates glucose transport in cardiomyocytes via a monoamine oxidase-dependent reaction. Biochem. J. 1995, 311 Pt 2, 575–583.
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351.
- Gibb, A.A.; Lorkiewicz, P.K.; Zheng, Y.T.; Zhang, X.; Bhatnagar, A.; Jones, S.P.; Hill, B.G. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem. J. 2017, 474, 2785–2801.
- Bianchi, P.; Pimentel, D.R.; Murphy, M.P.; Colucci, W.S.; Parini, A. A new hypertrophic mechanism of serotonin in cardiac myocytes: Receptor-independent ROS generation. FASEB J. 2005, 19, 641–643.
- Shao, W.; Shu, S.; Liu, R.; Jiang, Y.; Zhang, W.; Men, H. Monoamine oxidase inhibitors protect against coronary heart disease in rodent rat models: A pilot study. Pak. J. Pharm. Sci. 2019, 32, 371–375.
- Villeneuve, C.; Caudrillier, A.; Ordener, C.; Pizzinat, N.; Parini, A.; Mialet-Perez, J. Dose-dependent activation of distinct hypertrophic pathways by serotonin in cardiac cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H821–H828.
- Lairez, O.; Cognet, T.; Schaak, S.; Calise, D.; Guilbeau-Frugier, C.; Parini, A.; Mialet-Perez, J. Role of serotonin 5-HT2A receptors in the development of cardiac hypertrophy in response to aortic constriction in mice. J. Neural. Transm. (Vienna) 2013, 120, 927–935.
- Sommer, N.; Schulz, R. Mitochondrial Monoamine Oxidase: Another Player in Pulmonary Hypertension? Am. J. Respir. Cell Mol. Biol. 2021, 64, 277–278.
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332.
- Sonobe, T.; Akiyama, T.; Du, C.K.; Pearson, J.T. Serotonin uptake via plasma membrane monoamine transporter during myocardial ischemia-reperfusion in the rat heart in vivo. Physiol. Rep. 2019, 7, e14297.
- Du, C.K.; Zhan, D.Y.; Akiyama, T.; Inagaki, T.; Shishido, T.; Shirai, M.; Pearson, J.T. Myocardial interstitial levels of serotonin and its major metabolite 5-hydroxyindole acetic acid during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H60–H67.
- Inagaki, T.; Akiyama, T.; Du, C.K.; Zhan, D.Y.; Yoshimoto, M.; Shirai, M. Monoamine oxidase-induced hydroxyl radical production and cardiomyocyte injury during myocardial ischemia-reperfusion in rats. Free Radic. Res. 2016, 50, 645–653.
- Heger, J.; Hirschhauser, C.; Bornbaum, J.; Sydykov, A.; Dempfle, A.; Schneider, A.; Braun, T.; Schluter, K.D.; Schulz, R. Cardiomyocytes-specific deletion of monoamine oxidase B reduces irreversible myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 14–23.
- Bianchi, P.; Kunduzova, O.; Masini, E.; Cambon, C.; Bani, D.; Raimondi, L.; Seguelas, M.H.; Nistri, S.; Colucci, W.; Leducq, N.; et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 2005, 112, 3297–3305.
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49.
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174.
- Danila, M.D.; Privistirescu, A.I.; Mirica, S.N.; Sturza, A.; Ordodi, V.; Noveanu, L.; Duicu, O.M.; Muntean, D.M. Acute inhibition of monoamine oxidase and ischemic preconditioning in isolated rat hearts: Interference with postischemic functional recovery but no effect on infarct size reduction. Can. J. Physiol. Pharmacol. 2015, 93, 819–825.
- Varela, A.; Mavroidis, M.; Katsimpoulas, M.; Sfiroera, I.; Kappa, N.; Mesa, A.; Kostomitsopoulos, N.G.; Cokkinos, D.V. The neuroprotective agent Rasagiline mesylate attenuates cardiac remodeling after experimental myocardial infarction. ESC Heart Fail. 2017, 4, 331–340.
- Wang, Y.; Zhao, M.; Xu, B.; Bahriz, S.M.F.; Zhu, C.; Jovanovic, A.; Ni, H.; Jacobi, A.; Kaludercic, N.; Di Lisa, F.; et al. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular beta(1)AR signaling to calibrate cardiac contractile function. Basic Res. Cardiol. 2022, 117, 37.
- Wang, Y.; Zhao, M.; Shi, Q.; Xu, B.; Zhu, C.; Li, M.; Mir, V.; Bers, D.M.; Xiang, Y.K. Monoamine Oxidases Desensitize Intracellular beta(1)AR Signaling in Heart Failure. Circ. Res. 2021, 129, 965–967.
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid. Redox Signal. 2016, 25, 10–27.
- Martini, H.; Lefevre, L.; Sayir, S.; Itier, R.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Pizzinat, N.; Cussac, D.; et al. Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. Int. J. Mol. Sci. 2021, 22, 2245.
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69.
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492.
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabo, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 531–550.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
07 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No