 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Makoto Murakami | -- | 2753 | 2023-03-30 13:02:16 | | | |
| 2 | Lindsay Dong | Meta information modification | 2753 | 2023-03-31 07:44:14 | | | | |
| 3 | Lindsay Dong | + 1 word(s) | 2754 | 2023-03-31 09:41:53 | | |
Video Upload Options
The lysophospholipase PNPLA7 is a member of the Ca2+-independent phospholipase A2 (iPLA2) family, as a key regulator of the production of glycerophosphocholine (GPC), a precursor of endogenous choline, whose methyl groups are preferentially fluxed into the methionine cycle in the liver. PNPLA7 deficiency in mice markedly decreases hepatic GPC, choline, and several metabolites related to choline/methionine metabolism, leading to various symptoms reminiscent of methionine shortage. Overall metabolic alterations in the liver of Pnpla7-null mice in vivo largely recapitulate those in methionine-deprived hepatocytes in vitro. Reduction of the methyl donor S-adenosylmethionine (SAM) after methionine deprivation decreases the methylation of the PNPLA7 gene promoter, relieves PNPLA7 expression, and thereby increases GPC and choline levels, likely as a compensatory adaptation. In line with the view that SAM prevents the development of liver cancer, the expression of PNPLA7, as well as several enzymes in the choline/methionine metabolism, is reduced in human hepatocellular carcinoma.
1. Introduction
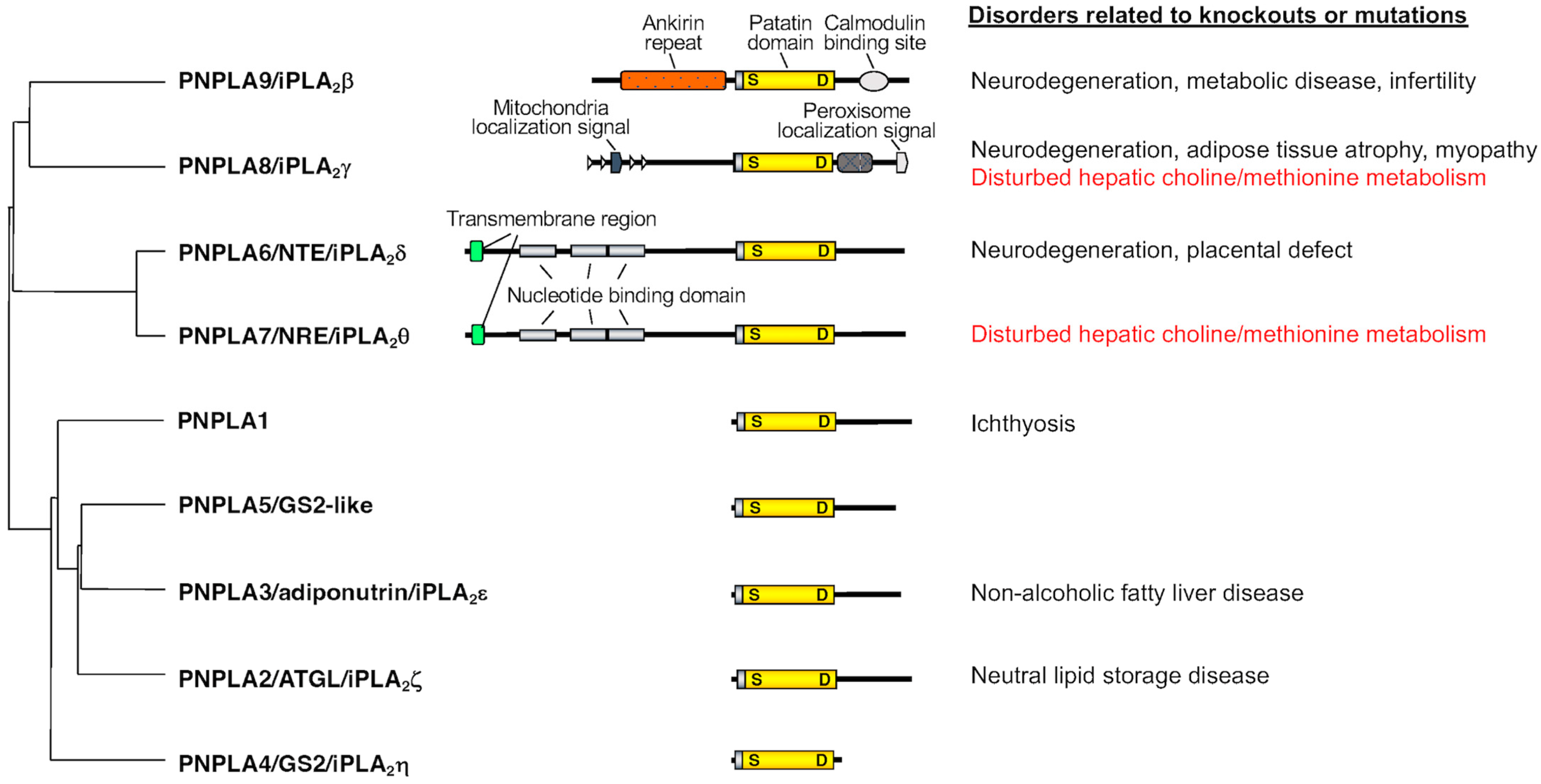
2. Properties of PNPLA7, the Closest Paralog of PNPLA6
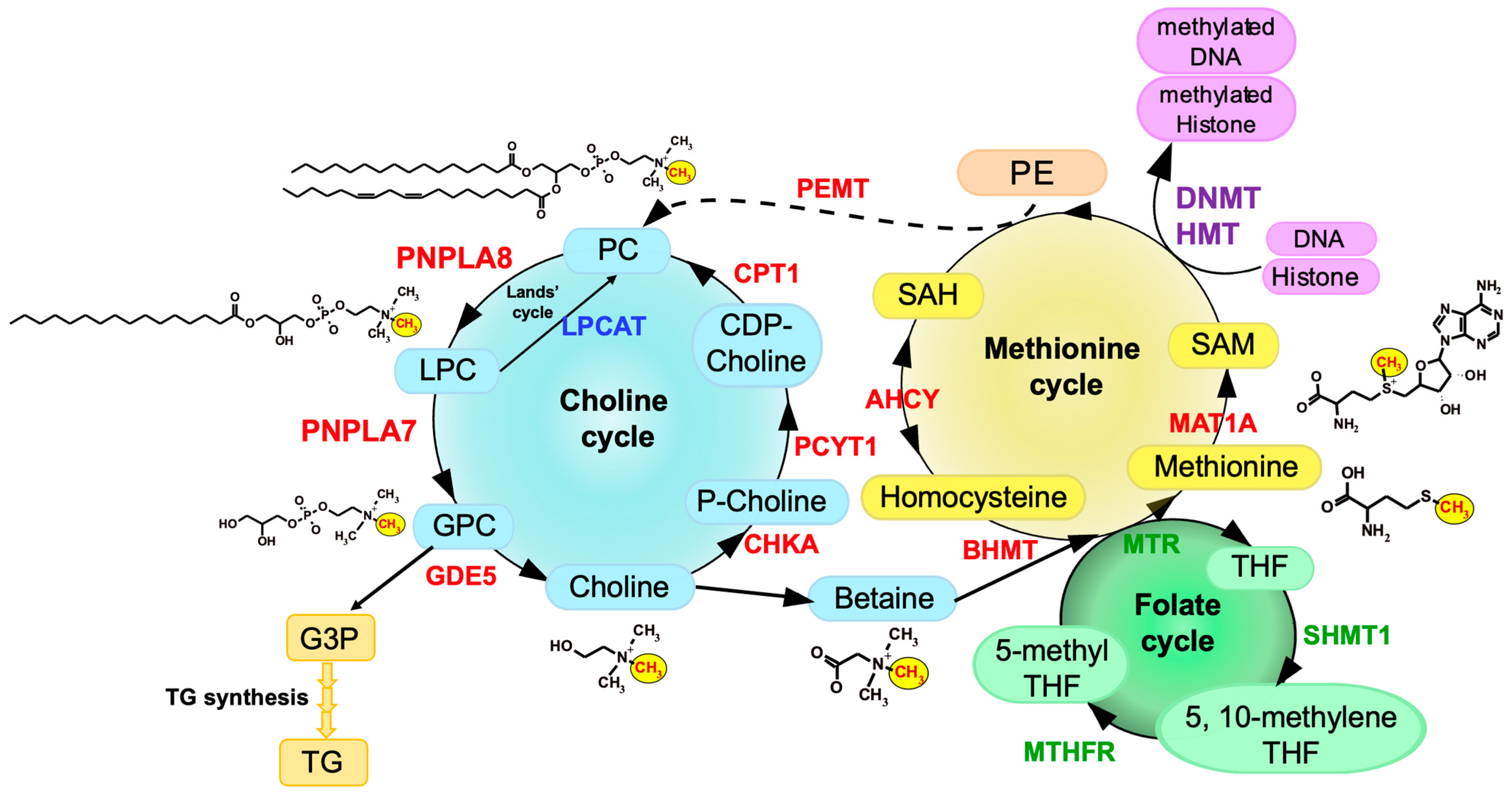
3. Choline/Methionine Metabolism
4. Phenotypes of Pnpla7-Deficient Mice
5. PNPLA8 Lies Upstream of PNPLA7
6. Regulation of PNPLA7 Expression in Hepatocytes by Methionine Availability
7. PNPLA7 Expression Is Decreased in Human Hepatocellular Carcinoma (HCC)
8. Other Lysophospholipases
References
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457.
- Kano, K.; Aoki, J.; Hla, T. Lysophospholipid mediators in health and disease. Ann. Rev. Pathol. 2022, 17, 459–483.
- Wang, A.; Dennis, E.A. Mammalian lysophospholipases. Biochim. Biophys. Acta 1999, 1439, 1–16.
- Lotti, M.; Moretto, A. Organophosphate-induced delayed polyneuropathy. Toxicol. Rev. 2005, 24, 37–49.
- Zaccheo, O.; Dinsdale, D.; Meacock, P.A.; Glynn, P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J. Biol. Chem. 2004, 279, 24024–24033.
- Muhlig-Versen, M.; da Cruz, A.B.; Tschape, J.A.; Moser, M.; Buttner, R.; Athenstaedt, K.; Glynn, P.; Kretzschmar, D. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J. Neurosci. 2005, 25, 2865–2873.
- Kretzschmar, D. PNPLA6/NTE, an evolutionary conserved phospholipase linked to a group of complex human diseases. Metabolites 2022, 12, 284.
- Kmoch, S.; Majewski, J.; Ramamurthy, V.; Cao, S.; Fahiminiya, S.; Ren, H.; MacDonald, I.M.; Lopez, I.; Sun, V.; Keser, V.; et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat. Commun. 2015, 6, 5614.
- Kienesberger, P.C.; Lass, A.; Preiss-Landl, K.; Wolinski, H.; Kohlwein, S.D.; Zimmermann, R.; Zechner, R. Identification of an insulin-regulated lysophospholipase with homology to neuropathy target esterase. J. Biol. Chem. 2008, 283, 5908–5917.
- Hirabayashi, T.; Kawaguchi, M.; Harada, S.; Mouri, M.; Takamiya, R.; Miki, Y.; Sato, H.; Taketomi, Y.; Yokoyama, K.; Kobayashi, T.; et al. Hepatic phosphatidylcholine catabolism driven by PNPLA7 and PNPLA8 supplies endogenous choline to replenish the methionine cycle with methyl groups. Cell Rep. 2023, 42, 111940.
- Heier, C.; Kien, B.; Huang, F.; Eichmann, T.O.; Xie, H.; Zechner, R.; Chang, P.A. The phospholipase PNPLA7 functions as a lysophosphatidylcholine hydrolase and interacts with lipid droplets through its catalytic domain. J. Biol. Chem. 2017, 292, 19087–19098.
- Wang, X.; Guo, M.; Wang, Q.; Wang, Q.; Zuo, S.; Zhang, X.; Tong, H.; Chen, J.; Wang, H.; Chen, X.; et al. The patatin-like phospholipase domain containing protein 7 facilitates VLDL secretion by modulating ApoE stability. Hepatology 2020, 72, 1569–1585.
- Sato, H.; Taketomi, Y.; Miki, Y.; Murase, R.; Yamamoto, K.; Murakami, M. Secreted Phospholipase PLA2G2D Contributes to metabolic health by mobilizing ω3 polyunsaturated fatty acids in WAT. Cell Rep. 2020, 31, 107579.
- Sato, H.; Taketomi, Y.; Ushida, A.; Isogai, Y.; Kojima, T.; Hirabayashi, T.; Miki, Y.; Yamamoto, K.; Nishito, Y.; Kobayashi, T.; et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 2014, 20, 119–132.
- Taketomi, Y.; Ueno, N.; Kojima, T.; Sato, H.; Murase, R.; Yamamoto, K.; Tanaka, S.; Sakanaka, M.; Nakamura, M.; Nishito, Y.; et al. Mast cell maturation is driven via a group III phospholipase A2-prostaglandin D2-DP1 receptor paracrine axis. Nat. Immunol. 2013, 14, 554–563.
- Yamamoto, K.; Miki, Y.; Sato, M.; Taketomi, Y.; Nishito, Y.; Taya, C.; Muramatsu, K.; Ikeda, K.; Nakanishi, H.; Taguchi, R.; et al. The role of group IIF-secreted phospholipase A2 in epidermal homeostasis and hyperplasia. J. Exp. Med. 2015, 212, 1901–1919.
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2013, 67, 615–623.
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572.
- Harayama, T.; Eto, M.; Shindou, H.; Kita, Y.; Otsubo, E.; Hishikawa, D.; Ishii, S.; Sakimura, K.; Mishina, M.; Shimizu, T. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metab. 2014, 20, 295–305.
- Shindou, H.; Hishikawa, D.; Harayama, T.; Yuki, K.; Shimizu, T. Recent progress on acyl CoA: Lysophospholipid acyltransferase research. J. Lipid Res. 2009, 50, S46–S51.
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637.
- Teng, Y.W.; Ellis, J.M.; Coleman, R.A.; Zeisel, S.H. Mouse betaine-homocysteine S-methyltransferase deficiency reduces body fat via increasing energy expenditure and impairing lipid synthesis and enhancing glucose oxidation in white adipose tissue. J. Biol. Chem. 2012, 287, 16187–16198.
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011, 34, 3–15.
- Cano, A.; Buque, X.; Martinez-Una, M.; Aurrekoetxea, I.; Menor, A.; Garcia-Rodriguez, J.L.; Lu, S.C.; Martinez-Chantar, M.L.; Mato, J.M.; Ochoa, B.; et al. Methionine adenosyltransferase 1A gene deletion disrupts hepatic very low-density lipoprotein assembly in mice. Hepatology 2011, 54, 1975–1986.
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26.
- Mato, J.M.; Martinez-Chantar, M.L.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189.
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542.
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Tu, B.P. A metabolic function for phospholipid and histone methylation. Mol. Cell 2017, 66, 180–193.e188.
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawlowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282.
- Gujar, H.; Weisenberger, D.J.; Liang, G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes 2019, 10, 172.
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010, 17, 2141–2151.
- Moghe, A.; Joshi-Barve, S.; Ghare, S.; Gobejishvili, L.; Kirpich, I.; McClain, C.J.; Barve, S. Histone modifications and alcohol-induced liver disease: Are altered nutrients the missing link? World J. Gastroenterol. 2010, 17, 2465–2472.
- Mancuso, D.J.; Sims, H.F.; Han, X.; Jenkins, C.M.; Guan, S.P.; Yang, K.; Moon, S.H.; Pietka, T.; Abumrad, N.A.; Schlesinger, P.H.; et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 2007, 282, 34611–34622.
- Mancuso, D.J.; Sims, H.F.; Yang, K.; Kiebish, M.A.; Su, X.; Jenkins, C.M.; Guan, S.; Moon, S.H.; Pietka, T.; Nassir, F.; et al. Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 2010, 285, 36495–36510.
- Yan, W.; Jenkins, C.M.; Han, X.; Mancuso, D.J.; Sims, H.F.; Yang, K.; Gross, R.W. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2γ: Identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J. Biol. Chem. 2005, 280, 26669–26679.
- Zhang, X.; Zhang, J.; Wang, R.; Guo, S.; Zhang, H.; Ma, Y.; Liu, Q.; Chu, H.; Xu, X.; Zhang, Y.; et al. Hypermethylation reduces the expression of PNPLA7 in hepatocellular carcinoma. Oncol. Lett. 2016, 12, 670–674.
- Lozano-Rosas, M.G.; Chavez, E.; Velasco-Loyden, G.; Dominguez-Lopez, M.; Martinez-Perez, L.; Chagoya De Sanchez, V. Diminished S-adenosylmethionine biosynthesis and its metabolism in a model of hepatocellular carcinoma is recuperated by an adenosine derivative. Cancer Biol. Ther. 2020, 21, 81–94.
- Pascale, R.M.; Feo, C.F.; Calvisi, D.F.; Feo, F. Deregulation of methionine metabolism as determinant of progression and prognosis of hepatocellular carcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 36.
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542.
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565.
- Moser, M.; Li, Y.; Vaupel, K.; Kretzschmar, D.; Kluge, R.; Glynn, P.; Buettner, R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol. Cell Biol. 2004, 24, 1667–1679.
- Akassoglou, K.; Malester, B.; Xu, J.; Tessarollo, L.; Rosenbluth, J.; Chao, M.V. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 5075–5080.
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785.
- Read, D.J.; Li, Y.; Chao, M.V.; Cavanagh, J.B.; Glynn, P. Neuropathy target esterase is required for adult vertebrate axon maintenance. J. Neurosci. 2009, 29, 11594–11600.
- Loo, R.W.; Conde-Frieboes, K.; Reynolds, L.J.; Dennis, E.A. Activation, inhibition, and regiospecificity of the lysophospholipase activity of the 85-kDa group IV cytosolic phospholipase A2. J. Biol. Chem. 1997, 272, 19214–19219.
- Ghomashchi, F.; Naika, G.S.; Bollinger, J.G.; Aloulou, A.; Lehr, M.; Leslie, C.C.; Gelb, M.H. Interfacial kinetic and binding properties of mammalian group IVB phospholipase A2 (cPLA2β) and comparison with the other cPLA2 isoforms. J. Biol. Chem. 2010, 285, 36100–36111.
- Yamashita, A.; Tanaka, K.; Kamata, R.; Kumazawa, T.; Suzuki, N.; Koga, H.; Waku, K.; Sugiura, T. Subcellular localization and lysophospholipase/transacylation activities of human group IVC phospholipase A2 (cPLA2γ). Biochim. Biophys. Acta 2009, 1791, 1011–1022.
- Lio, Y.C.; Dennis, E.A. Interfacial activation, lysophospholipase and transacylase activity of group VI Ca2+-independent phospholipase A2. Biochim. Biophys. Acta 1998, 1392, 320–332.
- Thomas, G.; Betters, J.L.; Lord, C.C.; Brown, A.L.; Marshall, S.; Ferguson, D.; Sawyer, J.; Davis, M.A.; Melchior, J.T.; Blume, L.C.; et al. The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome. Cell Rep. 2013, 5, 508–520.
- Zhao, S.; Mugabo, Y.; Ballentine, G.; Attane, C.; Iglesias, J.; Poursharifi, P.; Zhang, D.; Nguyen, T.A.; Erb, H.; Prentki, R.; et al. α/β-Hydrolase domain 6 deletion induces adipose browning and prevents obesity and type 2 diabetes. Cell Rep. 2016, 14, 2872–2888.
- Blankman, J.L.; Long, J.Z.; Trauger, S.A.; Siuzdak, G.; Cravatt, B.F. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad. Sci. USA 2013, 110, 1500–1505.
- Zhang, M.; Zhou, L.; Xu, Y.; Yang, M.; Komaniecki, G.P.; Kosciuk, T.; Chen, X.; Lu, X.; Zou, X.; Linder, M.E.; et al. A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 2020, 586, 434–439.

