4. Phenotypes of Pnpla7-Deficient Mice
Pnpla7−/− mice are born normally, yet they suffer from growth retardation after weaning and die within a few months
[10][14].
Pnpla7−/− mice exhibit kyphosis (excessive convex backward curvature of the spine) with muscle weakness and have markedly reduced visceral and subcutaneous fats, in which adipocytes contain smaller lipid droplets and more mitochondria and show some features of beige adipocytes with greater expression of thermogenic markers such as UCP1. In addition,
Pnpla7−/− mice display increases in food intake, locomotion, oxygen consumption, and dependence on lipid over carbohydrate as an energy source. The serum levels of glucose, TG, insulin and leptin are decreased, while the level of the ketone body β-hydroxybutyrate, a marker of starvation, is elevated, in
Pnpla7−/− mice. Although insulin sensitivity is unaltered, hepatic gluconeogenesis and VLDL secretion are compromised by PNPLA7 deficiency. Probably due to the lower levels of blood glucose and TG as fuels,
Pnpla7−/− mice have a lower body temperature.
Hepatic expression of genes related to lipogenesis is decreased, while that of genes related to lipolysis and fatty acid β-oxidation is elevated, in Pnpla7–/– mice. Furthermore, PNPLA7 deficiency markedly increases the blood level of the hepatokine FGF21, which could explain some of the metabolic phenotypes observed in Pnpla7−/− mice.
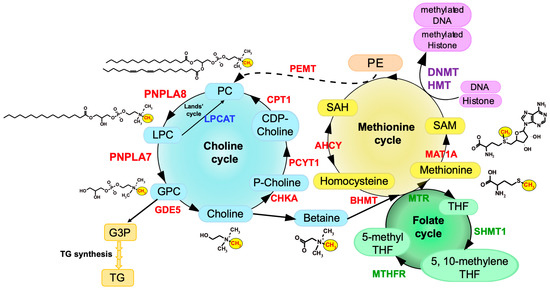
Metabolome analysis has revealed that the levels of GPC, choline and phosphocholine are markedly reduced, while those of LPC species are reciprocally elevated, in the liver of
Pnpla7−/− mice relative to
Pnpla7+/+ mice, indicating that PNPLA7 acts as a major lysophospholipase that hydrolyzes LPC to GPC in this tissue
[10][14]. In corroboration, acute knockdown of PNPLA7 in cultured hepatocytes decreases, whereas its overexpression conversely increases, cellular GPC levels. In contrast, PNPLA7 deficiency does not profoundly affect GPC levels in non-hepatic tissues such as adipose tissue, brain and kidney, where PNPLA6 may play a major or redundant role in GPC production. Notably, hepatic levels of a series of metabolites related to the methionine cycle such as betaine, methionine, and SAM, as well as the ratio of SAM/SAH (an indicator of the methylation flux), are markedly reduced in
Pnpla7−/− mice.
These results suggest that PNPLA7 deficiency reduces the flux of labile methyl groups from choline driven by the PC-catabolic pathway to the methionine cycle, in association with marked changes in the levels of related metabolites in the liver, thus underscoring a crucial role of PNPLA7 in the mobilization of hydrophilic choline from hydrophobic PC.
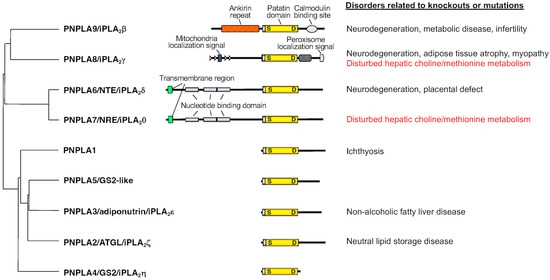
5. PNPLA8 Lies Upstream of PNPLA7
Of the knockout mouse strains for various PLA
2s examined so far
[1], only
Pnpla8−/− mice share several common features with
Pnpla7−/− mice. These phenotypes include reduced body weight and adiposity, muscle weakness leading to kyphosis, increased oxygen consumption, hypothermia, and reduced blood levels of glucose, TG, insulin, and leptin
[33][34][45,46]. Among the PLA
2 family, PNPLA8 shows the highest expression in mouse liver, with the magnitude order of PNPLA8 > PNPLA9 = PNPLA7 > PNPLA6 >> other PLA
2s. Importantly, as in
Pnpla7−/− mice, hepatic levels of GPC, choline, betaine, and SAM are substantially reduced in
Pnpla8−/− mice
[10][14]. Moreover, more than one third of the upregulated or downregulated genes, including those related to choline/methionine metabolism, fat synthesis and utilization, and glucose metabolism, in
Pnpla8−/− liver are commonly changed in
Pnpla7−/− liver. Hepatic expression of FGF21 is increased in both
Pnpla7−/− and
Pnpla8−/− mice. Thus, the phenotypes observed in
Pnpla8−/− mice are strikingly similar to those observed in
Pnpla7−/− mice, implying that PNPLA7 and PNPLA8 lie in the common metabolic pathway. Considering that PNPLA8 displays PLA
1/PLA
2 rather than lysophospholipase activity
[35][47], it is likely that PNPLA8 is a major upstream PLA
1/PLA
2 involved in the conversion of PC to LPC in the context of hepatic PC catabolism.
6. Regulation of PNPLA7 Expression in Hepatocytes by Methionine Availability
Pnpla7 expression is upregulated in the liver and adipose tissue of mice after fasting
[9][10][13,14]. Since PNPLA7 is functionally linked to choline/methionine metabolism, it is likely that its expression is regulated by some metabolites in this metabolic pathway. Indeed, PNPLA7 expression is uniquely regulated by the availability of methionine and thereby SAM, being unregulated by methionine shortage and downregulated by methionine supplementation. The promoter region of the
PNPLA7 gene is hypermethylated in several human hepatocellular carcinoma cell lines, including HepG2 and Huh7, and treatment of the cells with a DNMT inhibitor increases
PNPLA7 expression
[36][51]. Hepatocytes in methionine-free culture show a lower methylation level at a CpG site in the
PNPLA7 promoter than those in methionine-sufficient culture. Thus,
the decreased SAM flux in methionine-deprived cells results in reduced methylation of the PNPLA7 promoter, thereby allowing increased PNPLA7 expression.expression.
7. PNPLA7 Expression Is Decreased in Human Hepatocellular Carcinoma (HCC)
Current evidence suggests that SAM deficiency favors HCC development, which can be prevented by an exogenous supply of SAM
[37][38][57,58]. Indeed,
Mat1a-deficient mice, which display hepatic SAM deficiency, are highly susceptible to liver cancer, whereas hepatoma cells transfected with
MAT1A grow more slowly than control cells
[39][40][32,59]. In human HCC, the expression levels of
PNPLA7 and
PNPLA8 are markedly reduced in tumor tissues compared to non-tumor tissues, regardless of the stage or etiology of HCC. In addition to the expression levels of
PNPLA7 and
PNPLA8, those of enzymes in the methionine cycle (
BHMT, MAT1A, and
PEMT) are also lower in the tumors than in the controls.
Thus, multiple enzymes involved in PC catabolism and the methionine cycle are downregulated in HCC, implying that the reduced methyl group flux through this metabolic pathway contributes to progression of HCC. Thus, multiple enzymes involved in PC catabolism and the methionine cycle are downregulated in HCC, implying that the reduced methyl group flux through this metabolic pathway contributes to progression of HCC.
8. Other Lysophospholipases
Beyond PNPLA7, several enzymes in the PLA
2 family have been reported to possess lysophospholipase activity. As mentioned above, it is likely that PNPLA6 plays a compensatory or overlapping role with PNPLA7 in PC catabolism and recycling in tissue-specific contexts. Global
Pnpla6-deficient (
Pnpla6−/−) mice die in utero around E7.5 due to placental development
[41][65]. Brain-specific
Pnpla6-deficient mice display striking defects in motor coordination and neuronal degeneration with loss of Purkinje cells, disruption of the ER, accumulation of reticular aggregates, and an increase of PC
[42][66], resembling the features of PNPLA6-related disorders
[8][12] or hereditary spastic paraplegia
[43][67] in humans. In these mice, degeneration of the distal parts of the longest spinal axons becomes evident, with massive axon swelling in both the ascending and descending sensory and motor spinal tracts
[44][68], suggesting the PC/LPC catabolism driven by PNPLA6 in neurons may play a critical role in axonal membrane trafficking and secretion. All cPLA
2 enzymes (group IVA-F PLA
2s or cPLA
2α-ζ) and PNPLA9 (group VIA PLA
2 or iPLA
2β) display lysophospholipase activity in vitro
[45][46][47][48][69,70,71,72], yet the contribution of this activity to cPLA
2- or iPLA
2β-dependent biological processes remains unclear.
Among the ABHD family, ABHD6 appears to function as a lysophospholipase or monoacylglycerol lipase and is involved in the regulation of endocannabinoid signaling and metabolic disease [49][50]. ABHD12 hydrolyzes lysophosphatidylserine (LysoPS) as a LysoPS-specific lysophospholipase and its genetic deletion or mutation increases LysoPS in the brain, leading to symptoms reminiscent of PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts) [51]. Lastly, LYPLA1 and LYPLA2 are referred to as lysophospholipases, but it is now obvious that they act as acyl-thioesterases that catalyze the depalmitoylation of palmitoylated proteins [52].Among the ABHD family, ABHD6 appears to function as a lysophospholipase or monoacylglycerol lipase and is involved in the regulation of endocannabinoid signaling and metabolic disease [73,74]. ABHD12 hydrolyzes lysophosphatidylserine (LysoPS) as a LysoPS-specific lysophospholipase and its genetic deletion or mutation increases LysoPS in the brain, leading to symptoms reminiscent of PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts) [75]. Lastly, LYPLA1 and LYPLA2 are referred to as lysophospholipases, but it is now obvious that they act as acyl-thioesterases that catalyze the depalmitoylation of palmitoylated proteins [77].