The lysophospholipase PNPLA7 is a member of the Ca2+-independent phospholipase A2 (iPLA2) family, as a key regulator of the production of glycerophosphocholine (GPC), a precursor of endogenous choline, whose methyl groups are preferentially fluxed into the methionine cycle in the liver. PNPLA7 deficiency in mice markedly decreases hepatic GPC, choline, and several metabolites related to choline/methionine metabolism, leading to various symptoms reminiscent of methionine shortage. Overall metabolic alterations in the liver of Pnpla7-null mice in vivo largely recapitulate those in methionine-deprived hepatocytes in vitro. Reduction of the methyl donor S-adenosylmethionine (SAM) after methionine deprivation decreases the methylation of the PNPLA7 gene promoter, relieves PNPLA7 expression, and thereby increases GPC and choline levels, likely as a compensatory adaptation. In line with the view that SAM prevents the development of liver cancer, the expression of PNPLA7, as well as several enzymes in the choline/methionine metabolism, is reduced in human hepatocellular carcinoma.
- lysophospholipase
- phospholipase A2
- phosphatidylcholine
- lysophosphatidylcholine
- glycerophosphocholine
- S-adenosylmethionine
1. Introduction
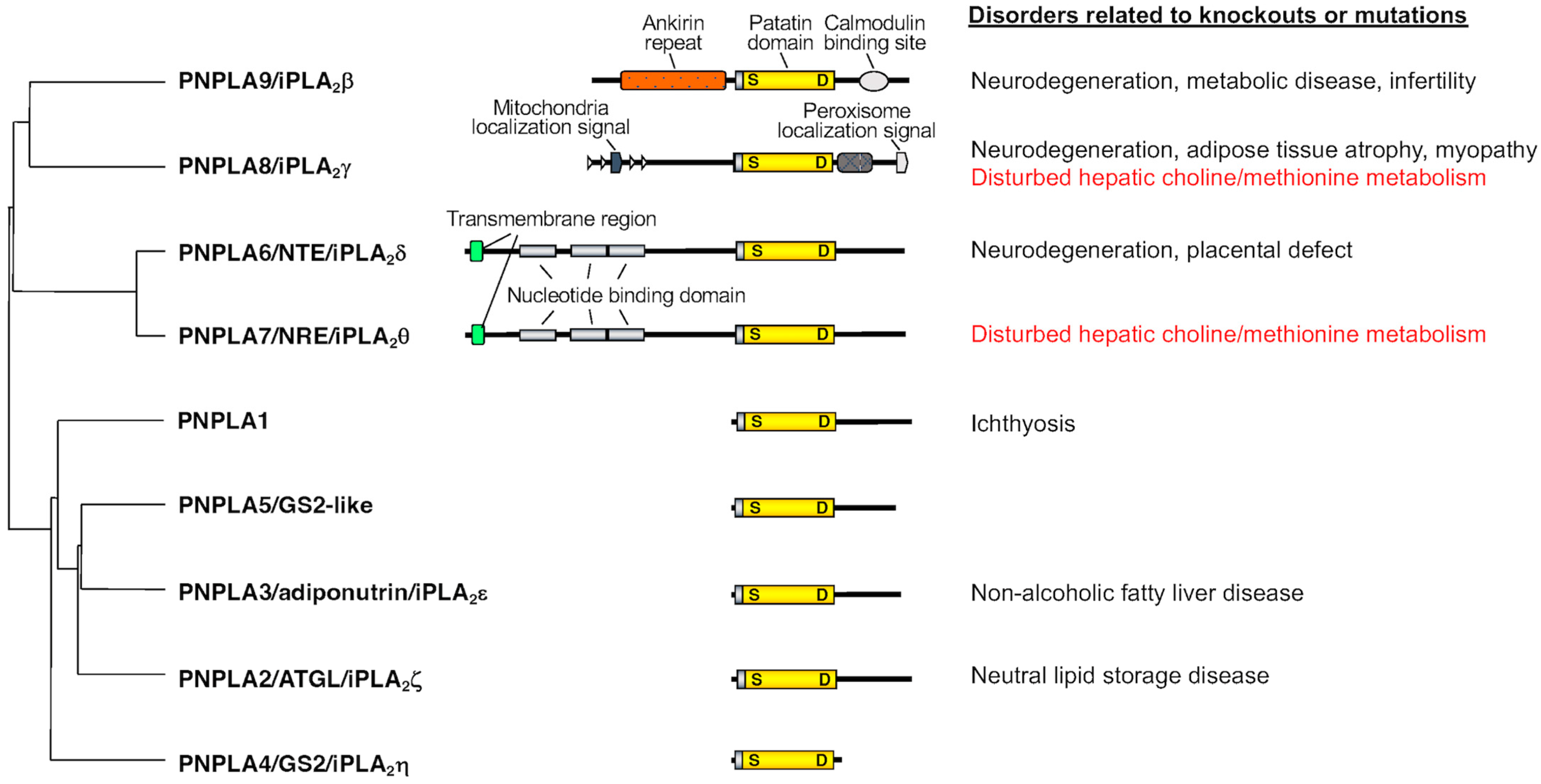
2. Properties of PNPLA7, the Closest Paralog of PNPLA6
PNPLA6, also known as neuropathy target esterase (NTE) or iPLA2δ, was originally identified in 1998 as a key factor responsible for organophosphate-induced delayed neuropathy (OPIDN), a neurodegenerative disorder that occurs within a few weeks after exposure to pesticides and nerve agents containing organophosphates [8][4]. PNPLA6 displays phospholipase B (PLA1/PLA2 + lysophospholipase) activity, hydrolyzing both the sn-1 and sn-2 fatty acyl chains of phosphatidylcholine (PC) and lysophosphatidylcholine (LPC) [9,10,11][5][6][7]. Mutations in the human PNPLA6 gene are linked to rare inherited diseases termed PNPLA6-related disorders, with clinical symptoms including spastic paraplegia, ataxia, hypogonadism, and chorioretinal dystrophy, reminiscent of the OPIDN pathology [12][8]. PNPLA7, also referred to as NTE-related esterase (NRE) or iPLA2θ, was identified in 2008 as the closest paralog of PNPLA6 [13][9]. The amino acid sequences of PNPLA6 and PNPLA7 are ~60% identical, with the N-terminal side facing the endoplasmic reticulum (ER) across the transmembrane domain and the C-terminal side including the catalytic patatin domain facing the cytosol (Figure 1). PNPLA7 displays lysophospholipase activity, hydrolyzing the sn-1 or sn-2 ester of LPC, although it does not catalyze the hydrolysis of PC via PLA1/PLA2 activity [13,14][9][10]. Both PNPLA6 and PNPLA7 are widely expressed in various tissues, with PNPLA6 being abundantly expressed in the central nervous system and PNPLA7 in metabolically active tissues including skeletal muscle, liver, and adipose tissue [14][10]. Expression of PNPLA7, but not PNPLA6, is upregulated in the liver and adipose tissue during fasting, suggesting its nutritional regulation [14][10]. In 3T3-L1 adipocytes, expression of PNPLA7 is downregulated by insulin [13][9]. Upon elevated fatty acid flux, the catalytic domain of PNPLA7 targets cellular lipid droplets in response to increased cAMP levels [15][11]. A recent study has reported that knockdown of hepatic PNPLA7 using adenoviral shRNA delivery impairs the secretion of very low-density lipoprotein (VLDL), proposing that PNPLA7 stabilizes ApoE through protein–protein interaction independent of its lysophospholipase activity [16][12]. In the ongoing efforts to clarify the biological roles of the PLA2 family by gene targeting [17[13][14][15][16],18,19,20], scholars have recently uncovered a previously unrecognized role of PNPLA7 as a bona fide lysophospholipase in the liver [14][10]. The main physiological role of the lysophospholipase PNPLA7 is not merely to remove otherwise toxic LPC when accumulated in excess or to modulate lysophospholipid mediator signaling, but rather to generate glycerophosphocholine (GPC), a hydrolytic product of LPC, as a precursor of free choline, whose labile methyl groups are then preferentially fluxed into the methionine cycle (Figure 2).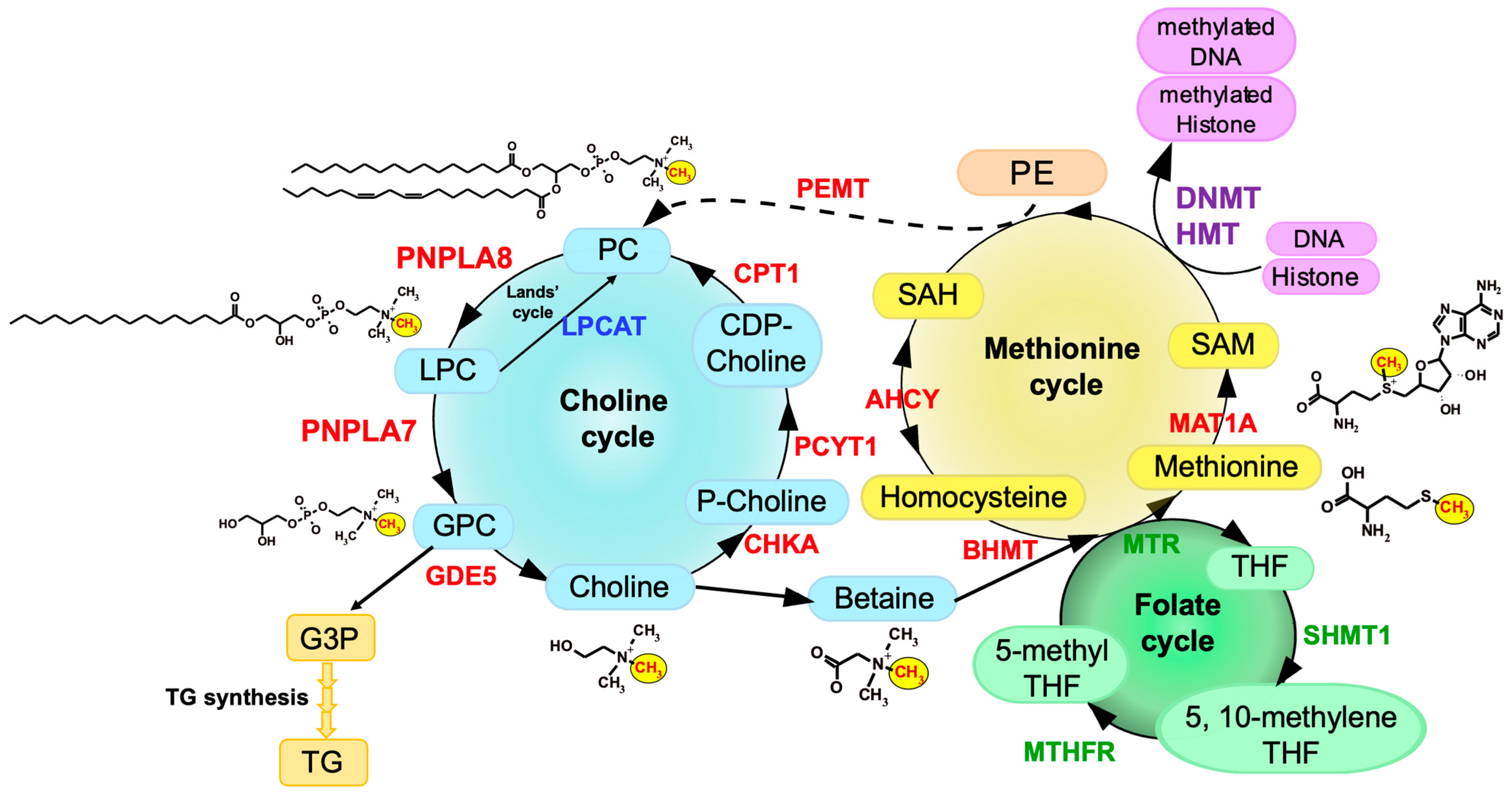
3. Choline/Methionine Metabolism
4. Phenotypes of Pnpla7-Deficient Mice
Pnpla7−/− mice are born normally, yet they suffer from growth retardation after weaning and die within a few months [14][10]. Pnpla7−/− mice exhibit kyphosis (excessive convex backward curvature of the spine) with muscle weakness and have markedly reduced visceral and subcutaneous fats, in which adipocytes contain smaller lipid droplets and more mitochondria and show some features of beige adipocytes with greater expression of thermogenic markers such as UCP1. In addition, Pnpla7−/− mice display increases in food intake, locomotion, oxygen consumption, and dependence on lipid over carbohydrate as an energy source. The serum levels of glucose, TG, insulin and leptin are decreased, while the level of the ketone body β-hydroxybutyrate, a marker of starvation, is elevated, in Pnpla7−/− mice. Although insulin sensitivity is unaltered, hepatic gluconeogenesis and VLDL secretion are compromised by PNPLA7 deficiency. Probably due to the lower levels of blood glucose and TG as fuels, Pnpla7−/− mice have a lower body temperature. Hepatic expression of genes related to lipogenesis is decreased, while that of genes related to lipolysis and fatty acid β-oxidation is elevated, in Pnpla7–/– mice. Furthermore, PNPLA7 deficiency markedly increases the blood level of the hepatokine FGF21, which could explain some of the metabolic phenotypes observed in Pnpla7−/− mice. Metabolome analysis has revealed that the levels of GPC, choline and phosphocholine are markedly reduced, while those of LPC species are reciprocally elevated, in the liver of Pnpla7−/− mice relative to Pnpla7+/+ mice, indicating that PNPLA7 acts as a major lysophospholipase that hydrolyzes LPC to GPC in this tissue [14][10]. In corroboration, acute knockdown of PNPLA7 in cultured hepatocytes decreases, whereas its overexpression conversely increases, cellular GPC levels. In contrast, PNPLA7 deficiency does not profoundly affect GPC levels in non-hepatic tissues such as adipose tissue, brain and kidney, where PNPLA6 may play a major or redundant role in GPC production. Notably, hepatic levels of a series of metabolites related to the methionine cycle such as betaine, methionine, and SAM, as well as the ratio of SAM/SAH (an indicator of the methylation flux), are markedly reduced in Pnpla7−/− mice. These results suggest that PNPLA7 deficiency reduces the flux of labile methyl groups from choline driven by the PC-catabolic pathway to the methionine cycle, in association with marked changes in the levels of related metabolites in the liver, thus underscoring a crucial role of PNPLA7 in the mobilization of hydrophilic choline from hydrophobic PC.5. PNPLA8 Lies Upstream of PNPLA7
Of the knockout mouse strains for various PLA2s examined so far [1], only Pnpla8−/− mice share several common features with Pnpla7−/− mice. These phenotypes include reduced body weight and adiposity, muscle weakness leading to kyphosis, increased oxygen consumption, hypothermia, and reduced blood levels of glucose, TG, insulin, and leptin [45,46][24][25]. Among the PLA2 family, PNPLA8 shows the highest expression in mouse liver, with the magnitude order of PNPLA8 > PNPLA9 = PNPLA7 > PNPLA6 >> other PLA2s. Importantly, as in Pnpla7−/− mice, hepatic levels of GPC, choline, betaine, and SAM are substantially reduced in Pnpla8−/− mice [14][10]. Moreover, more than one third of the upregulated or downregulated genes, including those related to choline/methionine metabolism, fat synthesis and utilization, and glucose metabolism, in Pnpla8−/− liver are commonly changed in Pnpla7−/− liver. Hepatic expression of FGF21 is increased in both Pnpla7−/− and Pnpla8−/− mice. Thus, the phenotypes observed in Pnpla8−/− mice are strikingly similar to those observed in Pnpla7−/− mice, implying that PNPLA7 and PNPLA8 lie in the common metabolic pathway. Considering that PNPLA8 displays PLA1/PLA2 rather than lysophospholipase activity [47][26], it is likely that PNPLA8 is a major upstream PLA1/PLA2 involved in the conversion of PC to LPC in the context of hepatic PC catabolism.6. Regulation of PNPLA7 Expression in Hepatocytes by Methionine Availability
Pnpla7 expression is upregulated in the liver and adipose tissue of mice after fasting [13,14][9][10]. Since PNPLA7 is functionally linked to choline/methionine metabolism, it is likely that its expression is regulated by some metabolites in this metabolic pathway. Indeed, PNPLA7 expression is uniquely regulated by the availability of methionine and thereby SAM, being unregulated by methionine shortage and downregulated by methionine supplementation. The promoter region of the PNPLA7 gene is hypermethylated in several human hepatocellular carcinoma cell lines, including HepG2 and Huh7, and treatment of the cells with a DNMT inhibitor increases PNPLA7 expression [51][27]. Hepatocytes in methionine-free culture show a lower methylation level at a CpG site in the PNPLA7 promoter than those in methionine-sufficient culture. Thus, the decreased SAM flux in methionine-deprived cells results in reduced methylation of the PNPLA7 promoter, thereby allowing increased PNPLA7 expression.7. PNPLA7 Expression Is Decreased in Human Hepatocellular Carcinoma (HCC)
Current evidence suggests that SAM deficiency favors HCC development, which can be prevented by an exogenous supply of SAM [57,58][28][29]. Indeed, Mat1a-deficient mice, which display hepatic SAM deficiency, are highly susceptible to liver cancer, whereas hepatoma cells transfected with MAT1A grow more slowly than control cells [32,59][30][31]. In human HCC, the expression levels of PNPLA7 and PNPLA8 are markedly reduced in tumor tissues compared to non-tumor tissues, regardless of the stage or etiology of HCC. In addition to the expression levels ofPNPLA7 and PNPLA8, those of enzymes in the methionine cycle (BHMT, MAT1A, and PEMT) are also lower in the tumors than in the controls. Thus, multiple enzymes involved in PC catabolism and the methionine cycle are downregulated in HCC, implying that the reduced methyl group flux through this metabolic pathway contributes to progression of HCC. Thus, multiple enzymes involved in PC catabolism and the methionine cycle are downregulated in HCC, implying that the reduced methyl group flux through this metabolic pathway contributes to progression of HCC.8. Other Lysophospholipases
References
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457.
- Kano, K.; Aoki, J.; Hla, T. Lysophospholipid mediators in health and disease. Ann. Rev. Pathol. 2022, 17, 459–483.
- Wang, A.; Dennis, E.A. Mammalian lysophospholipases. Biochim. Biophys. Acta 1999, 1439, 1–16.
- Lotti, M.; Moretto, A. Organophosphate-induced delayed polyneuropathy. Toxicol. Rev. 2005, 24, 37–49.
- Zaccheo, O.; Dinsdale, D.; Meacock, P.A.; Glynn, P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J. Biol. Chem. 2004, 279, 24024–24033.
- Muhlig-Versen, M.; da Cruz, A.B.; Tschape, J.A.; Moser, M.; Buttner, R.; Athenstaedt, K.; Glynn, P.; Kretzschmar, D. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J. Neurosci. 2005, 25, 2865–2873.
- Kretzschmar, D. PNPLA6/NTE, an evolutionary conserved phospholipase linked to a group of complex human diseases. Metabolites 2022, 12, 284.
- Kmoch, S.; Majewski, J.; Ramamurthy, V.; Cao, S.; Fahiminiya, S.; Ren, H.; MacDonald, I.M.; Lopez, I.; Sun, V.; Keser, V.; et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat. Commun. 2015, 6, 5614.
- Kienesberger, P.C.; Lass, A.; Preiss-Landl, K.; Wolinski, H.; Kohlwein, S.D.; Zimmermann, R.; Zechner, R. Identification of an insulin-regulated lysophospholipase with homology to neuropathy target esterase. J. Biol. Chem. 2008, 283, 5908–5917.
- Hirabayashi, T.; Kawaguchi, M.; Harada, S.; Mouri, M.; Takamiya, R.; Miki, Y.; Sato, H.; Taketomi, Y.; Yokoyama, K.; Kobayashi, T.; et al. Hepatic phosphatidylcholine catabolism driven by PNPLA7 and PNPLA8 supplies endogenous choline to replenish the methionine cycle with methyl groups. Cell Rep. 2023, 42, 111940.
- Heier, C.; Kien, B.; Huang, F.; Eichmann, T.O.; Xie, H.; Zechner, R.; Chang, P.A. The phospholipase PNPLA7 functions as a lysophosphatidylcholine hydrolase and interacts with lipid droplets through its catalytic domain. J. Biol. Chem. 2017, 292, 19087–19098.
- Wang, X.; Guo, M.; Wang, Q.; Wang, Q.; Zuo, S.; Zhang, X.; Tong, H.; Chen, J.; Wang, H.; Chen, X.; et al. The patatin-like phospholipase domain containing protein 7 facilitates VLDL secretion by modulating ApoE stability. Hepatology 2020, 72, 1569–1585.
- Sato, H.; Taketomi, Y.; Miki, Y.; Murase, R.; Yamamoto, K.; Murakami, M. Secreted Phospholipase PLA2G2D Contributes to metabolic health by mobilizing ω3 polyunsaturated fatty acids in WAT. Cell Rep. 2020, 31, 107579.
- Sato, H.; Taketomi, Y.; Ushida, A.; Isogai, Y.; Kojima, T.; Hirabayashi, T.; Miki, Y.; Yamamoto, K.; Nishito, Y.; Kobayashi, T.; et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 2014, 20, 119–132.
- Taketomi, Y.; Ueno, N.; Kojima, T.; Sato, H.; Murase, R.; Yamamoto, K.; Tanaka, S.; Sakanaka, M.; Nakamura, M.; Nishito, Y.; et al. Mast cell maturation is driven via a group III phospholipase A2-prostaglandin D2-DP1 receptor paracrine axis. Nat. Immunol. 2013, 14, 554–563.
- Yamamoto, K.; Miki, Y.; Sato, M.; Taketomi, Y.; Nishito, Y.; Taya, C.; Muramatsu, K.; Ikeda, K.; Nakanishi, H.; Taguchi, R.; et al. The role of group IIF-secreted phospholipase A2 in epidermal homeostasis and hyperplasia. J. Exp. Med. 2015, 212, 1901–1919.
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2013, 67, 615–623.
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572.
- Harayama, T.; Eto, M.; Shindou, H.; Kita, Y.; Otsubo, E.; Hishikawa, D.; Ishii, S.; Sakimura, K.; Mishina, M.; Shimizu, T. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metab. 2014, 20, 295–305.
- Shindou, H.; Hishikawa, D.; Harayama, T.; Yuki, K.; Shimizu, T. Recent progress on acyl CoA: Lysophospholipid acyltransferase research. J. Lipid Res. 2009, 50, S46–S51.
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637.
- Teng, Y.W.; Ellis, J.M.; Coleman, R.A.; Zeisel, S.H. Mouse betaine-homocysteine S-methyltransferase deficiency reduces body fat via increasing energy expenditure and impairing lipid synthesis and enhancing glucose oxidation in white adipose tissue. J. Biol. Chem. 2012, 287, 16187–16198.
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011, 34, 3–15.
- Mancuso, D.J.; Sims, H.F.; Han, X.; Jenkins, C.M.; Guan, S.P.; Yang, K.; Moon, S.H.; Pietka, T.; Abumrad, N.A.; Schlesinger, P.H.; et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 2007, 282, 34611–34622.
- Mancuso, D.J.; Sims, H.F.; Yang, K.; Kiebish, M.A.; Su, X.; Jenkins, C.M.; Guan, S.; Moon, S.H.; Pietka, T.; Nassir, F.; et al. Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 2010, 285, 36495–36510.
- Yan, W.; Jenkins, C.M.; Han, X.; Mancuso, D.J.; Sims, H.F.; Yang, K.; Gross, R.W. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2γ: Identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J. Biol. Chem. 2005, 280, 26669–26679.
- Zhang, X.; Zhang, J.; Wang, R.; Guo, S.; Zhang, H.; Ma, Y.; Liu, Q.; Chu, H.; Xu, X.; Zhang, Y.; et al. Hypermethylation reduces the expression of PNPLA7 in hepatocellular carcinoma. Oncol. Lett. 2016, 12, 670–674.
- Lozano-Rosas, M.G.; Chavez, E.; Velasco-Loyden, G.; Dominguez-Lopez, M.; Martinez-Perez, L.; Chagoya De Sanchez, V. Diminished S-adenosylmethionine biosynthesis and its metabolism in a model of hepatocellular carcinoma is recuperated by an adenosine derivative. Cancer Biol. Ther. 2020, 21, 81–94.
- Pascale, R.M.; Feo, C.F.; Calvisi, D.F.; Feo, F. Deregulation of methionine metabolism as determinant of progression and prognosis of hepatocellular carcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 36.
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542.
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565.
- Moser, M.; Li, Y.; Vaupel, K.; Kretzschmar, D.; Kluge, R.; Glynn, P.; Buettner, R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol. Cell Biol. 2004, 24, 1667–1679.
- Akassoglou, K.; Malester, B.; Xu, J.; Tessarollo, L.; Rosenbluth, J.; Chao, M.V. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 5075–5080.
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785.
- Read, D.J.; Li, Y.; Chao, M.V.; Cavanagh, J.B.; Glynn, P. Neuropathy target esterase is required for adult vertebrate axon maintenance. J. Neurosci. 2009, 29, 11594–11600.
- Loo, R.W.; Conde-Frieboes, K.; Reynolds, L.J.; Dennis, E.A. Activation, inhibition, and regiospecificity of the lysophospholipase activity of the 85-kDa group IV cytosolic phospholipase A2. J. Biol. Chem. 1997, 272, 19214–19219.
- Ghomashchi, F.; Naika, G.S.; Bollinger, J.G.; Aloulou, A.; Lehr, M.; Leslie, C.C.; Gelb, M.H. Interfacial kinetic and binding properties of mammalian group IVB phospholipase A2 (cPLA2β) and comparison with the other cPLA2 isoforms. J. Biol. Chem. 2010, 285, 36100–36111.
- Yamashita, A.; Tanaka, K.; Kamata, R.; Kumazawa, T.; Suzuki, N.; Koga, H.; Waku, K.; Sugiura, T. Subcellular localization and lysophospholipase/transacylation activities of human group IVC phospholipase A2 (cPLA2γ). Biochim. Biophys. Acta 2009, 1791, 1011–1022.
- Lio, Y.C.; Dennis, E.A. Interfacial activation, lysophospholipase and transacylase activity of group VI Ca2+-independent phospholipase A2. Biochim. Biophys. Acta 1998, 1392, 320–332.