 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chunyuan Yin | -- | 2100 | 2023-03-20 15:40:26 | | | |
| 2 | Jessie Wu | Meta information modification | 2100 | 2023-03-21 02:07:01 | | | | |
| 3 | Jessie Wu | + 3 word(s) | 2103 | 2023-03-21 02:08:42 | | |
Video Upload Options
Alzheimer’s disease (AD) is an aging-related neurodegenerative disease, leading to the progressive loss of memory and other cognitive functions. Metabolomics allows the study of biochemical alterations in pathological processes which may be involved in AD progression and to discover new therapeutic targets. Metabolites are substrates, intermediates, and products of metabolic body processes, which typically are small molecules with a molecular weight of less than ~1.5 kDa. Since low molecular weight metabolites are intermediates or end products of cellular metabolism, metabolomics, or the study of metabolism can be considered one of the core disciplines of systems biology. It can help in improving our understanding of changes in biochemical pathways, revealing crucial information that is closely related to human disease or therapeutic status.
1. Arginine Metabolism
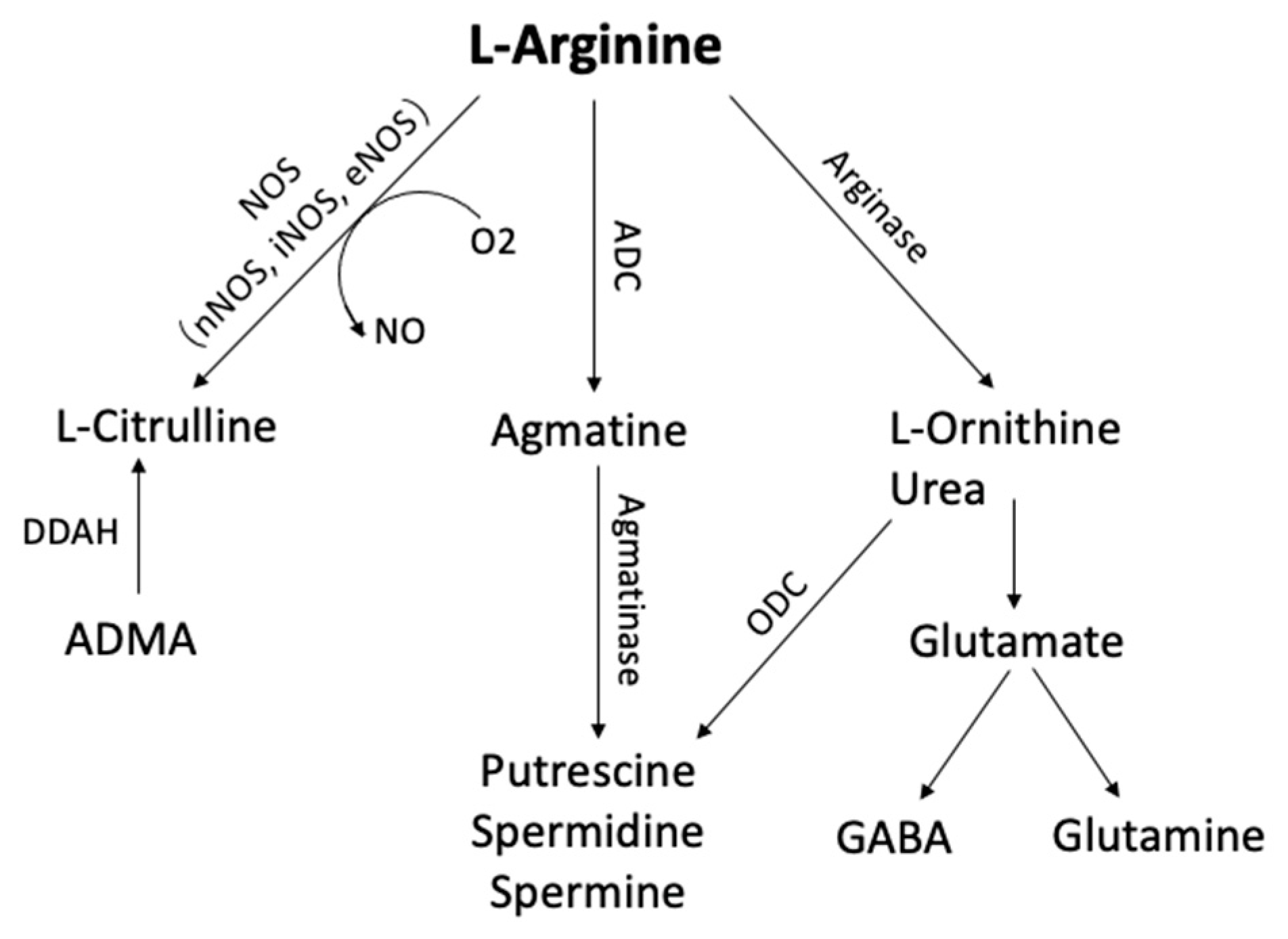
Figure 1. Arginine metabolic pathways. L-arginine can be metabolized by phosphatidic acid (PA), nitric oxide synthase (NOS), arginase, and arginine decarboxylase (ADC) to form several bioactive molecules. (ADC, Arginine decarboxylase; ADMA, NG-dimethyl-L-arginine; DDAH, dimethylarginine dimethylaminohydrolase; GABA, γ-aminobutyric acid; ODC, ornithine decarboxylase)
2. Alanine, Aspartate, and Glutamate Metabolism
3. Purine Metabolism
4. Taurine and Hypotaurine Metabolism
5. Cholinergic System
6. Fatty Acids
7. Glycerolipids
8. Glycerophospholipids
9. Sphingolipids
10. Cholesterol and Cholesteryl Esters
References
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336 Pt 1, 1–17.
- Morris, S.M., Jr. Enzymes of Arginine Metabolism. J. Nutr. 2004, 134, 2743S–2747S.
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653.
- Ellison, D.W.; Beal, M.F.; Mazurek, M.F.; Bird, E.D.; Martin, J.B. A postmortem study of amino acid neurotransmitters in Alzheimer’s disease. Ann. Neurol. 1986, 20, 616–621.
- Tumani, H.; Shen, G.; Peter, J.B.; Brück, W. Glutamine synthetase in cerebrospinal fluid, serum, and brain: A diagnostic marker for Alzheimer disease? Arch. Neurol. 1999, 56, 1241–1246.
- Satriano, J. Arginine pathways and the inflammatory response: Interregulation of nitric oxide and polyamines: Review article. Amino Acids 2004, 26, 321–329.
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230.
- Susswein, A.J.; Katzoff, A.; Miller, N.; Hurwitz, I. Nitric oxide and memory. Neuroscientist 2004, 10, 153–162.
- Feil, R.; Kleppisch, T. NO/cGMP-dependent modulation of synaptic transmission. In Pharmacology of Neurotransmitter Release; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 529–560.
- Böger, R.H.; Bode-Böger, S.M.; Frölich, J.C. The L-arginine-nitric oxide pathway: Role in atherosclerosis and therapeutic implications. Atherosclerosis 1996, 127, 1–11.
- Cooke, J.P.; Dzau, V.J. Nitric oxide synthase: Role in the genesis of vascular disease. Annu. Rev. Med. 1997, 48, 489–509.
- Cooke, J.P. The pivotal role of nitric oxide for vascular health. Can. J. Cardiol. 2004, 20 (Suppl. B), 7b–15b.
- Li, X.A.; Everson, W.; Smart, E.J. Nitric oxide, caveolae, and vascular pathology. Cardiovasc. Toxicol. 2006, 6, 1–13.
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric oxide and atherosclerosis: An update. Nitric Oxide 2006, 15, 265–279.
- Fonnum, F. Glutamate: A neurotransmitter in mammalian brain. J. Neurochem. 1984, 42, 1–11.
- Francis, P.T.; Sims, N.R.; Procter, A.W.; Bowen, D.M. Cortical pyramidal neurone loss may cause glutamatergic hypoactivity and cognitive impairment in Alzheimer’s disease: Investigative and therapeutic perspectives. J. Neurochem. 1993, 60, 1589–1604.
- Bruno, V.; Battaglia, G.; Copani, A.; D’Onofrio, M.; Di Iorio, P.; De Blasi, A.; Melchiorri, D.; Flor, P.J.; Nicoletti, F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033.
- Gadea, A.; López-Colomé, A.M. Glial transporters for glutamate, glycine and GABA I. Glutamate transporters. J. Neurosci. Res. 2001, 63, 453–460.
- Ozawa, S.; Kamiya, H.; Tsuzuki, K. Glutamate receptors in the mammalian central nervous system. Prog. Neurobiol. 1998, 54, 581–618.
- Lynch, G. Memory and the brain: Unexpected chemistries and a new pharmacology. Neurobiol. Learn. Mem. 1998, 70, 82–100.
- Riedel, G.; Micheau, J. Function of the hippocampus in memory formation: Desperately seeking resolution. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 835–853.
- Myhrer, T. Effects of selective perirhinal and postrhinal lesions on acquisition and retention of a visual discrimination task in rats. Neurobiol. Learn. Mem. 2000, 73, 68–78.
- Baudry, M.; Lynch, G. Remembrance of arguments past: How well is the glutamate receptor hypothesis of LTP holding up after 20 years? Neurobiol. Learn. Mem. 2001, 76, 284–297.
- Jay, T.M.; Zilkha, E.; Obrenovitch, T.P. Long-term potentiation in the dentate gyrus is not linked to increased extracellular glutamate concentration. J. Neurophysiol. 1999, 81, 1741–1748.
- Scannevin, R.H.; Huganir, R.L. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 2000, 1, 133–141.
- Ansoleaga, B.; Jové, M.; Schlüter, A.; Garcia-Esparcia, P.; Moreno, J.; Pujol, A.; Pamplona, R.; Portero-Otín, M.; Ferrer, I. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 68–80.
- Ipata, P.L.; Camici, M.; Micheli, V.; Tozz, M.G. Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 2011, 11, 909–922.
- Ferreira, I.L.; Resende, R.; Ferreiro, E.; Rego, A.C.; Pereira, C.F. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 2010, 11, 1193–1206.
- Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 2009, 41, 425–431.
- Lovell, M.A.; Markesbery, W.R. Oxidatively modified RNA in mild cognitive impairment. Neurobiol. Dis. 2008, 29, 169–175.
- Lovell, M.A.; Soman, S.; Bradley, M.A. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech. Ageing Dev. 2011, 132, 443–448.
- Markesbery, W.R.; Lovell, M.A. DNA oxidation in Alzheimer’s disease. Antioxid. Redox Signal. 2006, 8, 2039–2045.
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W., Jr.; Tabaton, M.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241.
- Kaddurah-Daouk, R.; Rozen, S.; Matson, W.; Han, X.; Hulette, C.M.; Burke, J.R.; Doraiswamy, P.M.; Welsh-Bohmer, K.A. Metabolomic changes in autopsy-confirmed Alzheimer’s disease. Alzheimers Dement. 2011, 7, 309–317.
- Isobe, C.; Abe, T.; Terayama, Y. Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2’-deoxyguanosine in the CSF of patients with Alzheimer’s disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. J. Neurol. 2010, 257, 399–404.
- Kaddurah-Daouk, R.; Zhu, H.; Sharma, S.; Bogdanov, M.; Rozen, S.G.; Matson, W.; Oki, N.O.; Motsinger-Reif, A.A.; Churchill, E.; Lei, Z.; et al. Alterations in metabolic pathways and networks in Alzheimer’s disease. Transl. Psychiatry 2013, 3, e244.
- Jové, M.; Portero-Otín, M.; Naudí, A.; Ferrer, I.; Pamplona, R. Metabolomics of human brain aging and age-related neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2014, 73, 640–657.
- Frosini, M.; Sesti, C.; Saponara, S.; Ricci, L.; Valoti, M.; Palmi, M.; Machetti, F.; Sgaragli, G. A specific taurine recognition site in the rabbit brain is responsible for taurine effects on thermoregulation. Br. J. Pharmacol. 2003, 139, 487–494.
- Bhat, M.A.; Ahmad, K.; Khan, M.S.A.; Bhat, M.A.; Almatroudi, A.; Rahman, S.; Jan, A.T. Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases. Biomolecules 2020, 10, 863.
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Abraham, J.R.; Apostolopoulos, V.; Zulli, A. The Anti-Inflammatory Effect of Taurine on Cardiovascular Disease. Nutrients 2020, 12, 2847.
- Schaffer, S.W.; Azuma, J.; Mozaffari, M. Role of antioxidant activity of taurine in diabetes. Can. J. Physiol. Pharmacol. 2009, 87, 91–99.
- Schaffer, S.; Takahashi, K.; Azuma, J. Role of osmoregulation in the actions of taurine. Amino Acids 2000, 19, 527–546.
- Foos, T.M.; Wu, J.Y. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem. Res. 2002, 27, 21–26.
- Vohra, B.P.; Hui, X. Improvement of impaired memory in mice by taurine. Neural Plast. 2000, 7, 245–259.
- Su, Y.; Fan, W.; Ma, Z.; Wen, X.; Wang, W.; Wu, Q.; Huang, H. Taurine improves functional and histological outcomes and reduces inflammation in traumatic brain injury. Neuroscience 2014, 266, 56–65.
- Malcangio, M.; Bartolini, A.; Ghelardini, C.; Bennardini, F.; Malmberg-Aiello, P.; Franconi, F.; Giotti, A. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology 1989, 98, 316–320.
- Javed, H.; Khan, A.; Vaibhav, K.; Moshahid Khan, M.; Ahmad, A.; Ejaz Ahmad, M.; Ahmad, A.; Tabassum, R.; Islam, F.; Safhi, M.M.; et al. Taurine ameliorates neurobehavioral, neurochemical and immunohistochemical changes in sporadic dementia of Alzheimer’s type (SDAT) caused by intracerebroventricular streptozotocin in rats. Neurol. Sci. 2013, 34, 2181–2192.
- Santa-María, I.; Hernández, F.; Moreno, F.J.; Avila, J. Taurine, an inducer for tau polymerization and a weak inhibitor for amyloid-beta-peptide aggregation. Neurosci. Lett. 2007, 429, 91–94.
- Pan, C.; Prentice, H.; Price, A.L.; Wu, J.Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids 2012, 43, 845–855.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115.
- Kása, P.; Rakonczay, Z.; Gulya, K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1997, 52, 511–535.
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933.
- de Carvalho, C.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583.
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505.
- Fonteh, A.N.; Cipolla, M.; Chiang, A.J.; Edminster, S.P.; Arakaki, X.; Harrington, M.G. Polyunsaturated Fatty Acid Composition of Cerebrospinal Fluid Fractions Shows Their Contribution to Cognitive Resilience of a Pre-symptomatic Alzheimer’s Disease Cohort. Front. Physiol. 2020, 11, 83.
- Cunnane, S.C.; Schneider, J.A.; Tangney, C.; Tremblay-Mercier, J.; Fortier, M.; Bennett, D.A.; Morris, M.C. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 691–697.
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Using direct infusion mass spectrometry for serum metabolomics in Alzheimer’s disease. Anal. Bioanal. Chem. 2014, 406, 7137–7148.
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann. Neurol. 1997, 42, 776–782.
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N.; Schneider, J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 940–946.
- Huang, T.L.; Zandi, P.P.; Tucker, K.L.; Fitzpatrick, A.L.; Kuller, L.H.; Fried, L.P.; Burke, G.L.; Carlson, M.C. Benefits of fatty fish on dementia risk are stronger for those without APOE epsilon4. Neurology 2005, 65, 1409–1414.
- Sanchez-Mejia, R.O.; Mucke, L. Phospholipase A2 and arachidonic acid in Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 784–790.
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266.
- Prasad, M.R.; Lovell, M.A.; Yatin, M.; Dhillon, H.; Markesbery, W.R. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 1998, 23, 81–88.
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimers Dement. 2017, 13, 140–151.
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688.
- Wood, P.L.; Medicherla, S.; Sheikh, N.; Terry, B.; Phillipps, A.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Targeted Lipidomics of Fontal Cortex and Plasma Diacylglycerols (DAG) in Mild Cognitive Impairment and Alzheimer’s Disease: Validation of DAG Accumulation Early in the Pathophysiology of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 537–546.
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 2015, 27, 270–278.
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29.
- Bargui, R.; Solgadi, A.; Prost, B.; Chester, M.; Ferreiro, A.; Piquereau, J.; Moulin, M. Phospholipids: Identification and Implication in Muscle Pathophysiology. Int. J. Mol. Sci. 2021, 22, 8176.
- Pettegrew, J.W.; Panchalingam, K.; Hamilton, R.L.; McClure, R.J. Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 2001, 26, 771–782.
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimers Dis. 2011, 24, 507–517.
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180.
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747.
- Wells, K.; Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neural membrane phospholipids in Alzheimer disease. Neurochem. Res. 1995, 20, 1329–1333.
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 1991, 26, 421–425.
- Stephenson, D.T.; Lemere, C.A.; Selkoe, D.J.; Clemens, J.A. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer’s disease brain. Neurobiol. Dis. 1996, 3, 51–63.
- Gattaz, W.F.; Maras, A.; Cairns, N.J.; Levy, R.; Förstl, H. Decreased phospholipase A2 activity in Alzheimer brains. Biol. Psychatry 1995, 37, 13–17.
- Schaeffer, E.L.; Gattaz, W.F. Requirement of hippocampal phospholipase A2 activity for long-term memory retrieval in rats. J. Neural Transm. 2007, 114, 379–385.
- Kim, H.Y.; Huang, B.X.; Spector, A.A. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 2014, 56, 1–18.
- Zhang, Y.Y.; Yang, L.Q.; Guo, L.M. Effect of phosphatidylserine on memory in patients and rats with Alzheimer’s disease. Genet. Mol. Res. 2015, 14, 9325–9333.
- Kidd, P.M. Omega-3 DHA and EPA for cognition, behavior, and mood: Clinical findings and structural-functional synergies with cell membrane phospholipids. Altern. Med. Rev. 2007, 12, 207–227.
- Frisardi, V.; Panza, F.; Seripa, D.; Farooqui, T.; Farooqui, A.A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 2011, 50, 313–330.
- Schaeffer, E.L.; da Silva, E.R.; Novaes Bde, A.; Skaf, H.D.; Gattaz, W.F. Differential roles of phospholipases A2 in neuronal death and neurogenesis: Implications for Alzheimer disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1381–1389.
- Perttu, E.K.; Kohli, A.G.; Szoka, F.C., Jr. Inverse-phosphocholine lipids: A remix of a common phospholipid. J. Am. Chem. Soc. 2012, 134, 4485–4488.
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; García-Gómez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278.
- Kang, S.; Han, J.; Song, S.Y.; Kim, W.S.; Shin, S.; Kim, J.H.; Ahn, H.; Jeong, J.H.; Hwang, S.J.; Sung, J.H. Lysophosphatidic acid increases the proliferation and migration of adipose-derived stem cells via the generation of reactive oxygen species. Mol. Med. Rep. 2015, 12, 5203–5210.
- Ahmad, S.; Orellana, A.; Kohler, I.; Frölich, L.; de Rojas, I.; Gil, S.; Boada, M.; Hernández, I.; Hausner, L.; Bakker, M.H.M.; et al. Association of lysophosphatidic acids with cerebrospinal fluid biomarkers and progression to Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 124.
- Jones, E.E.; Dworski, S.; Canals, D.; Casas, J.; Fabrias, G.; Schoenling, D.; Levade, T.; Denlinger, C.; Hannun, Y.A.; Medin, J.A.; et al. On-tissue localization of ceramides and other sphingolipids by MALDI mass spectrometry imaging. Anal. Chem. 2014, 86, 8303–8311.
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 346783.
- Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and sphingomyelinases in senile plaques. Neurobiol. Dis. 2014, 65, 193–201.
- Mielke, M.M.; Bandaru, V.V.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum ceramides increase the risk of Alzheimer disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641.
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial expression of ceramide in Alzheimer’s disease brains: A role during neuronal apoptosis. Neuroscience 2005, 130, 657–666.
- Couttas, T.A.; Kain, N.; Daniels, B.; Lim, X.Y.; Shepherd, C.; Kril, J.; Pickford, R.; Li, H.; Garner, B.; Don, A.S. Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2014, 2, 9.
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408.
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372.
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075.
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Björkhem, I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 2004, 45, 186–193.
- Mori, T.; Paris, D.; Town, T.; Rojiani, A.M.; Sparks, D.L.; Delledonne, A.; Crawford, F.; Abdullah, L.I.; Humphrey, J.A.; Dickson, D.W.; et al. Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J. Neuropathol. Exp. Neurol. 2001, 60, 778–785.

