Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matteo Pavan | -- | 4677 | 2023-03-07 11:31:01 | | | |
| 2 | Camila Xu | Meta information modification | 4677 | 2023-03-08 01:25:25 | | | | |
| 3 | Camila Xu | -65 word(s) | 4612 | 2023-03-08 01:33:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pavan, M.; Moro, S. Tackling Pandemics through Computer-Aided Drug Discovery Approaches. Encyclopedia. Available online: https://encyclopedia.pub/entry/41939 (accessed on 28 July 2026).
Pavan M, Moro S. Tackling Pandemics through Computer-Aided Drug Discovery Approaches. Encyclopedia. Available at: https://encyclopedia.pub/entry/41939. Accessed July 28, 2026.
Pavan, Matteo, Stefano Moro. "Tackling Pandemics through Computer-Aided Drug Discovery Approaches" Encyclopedia, https://encyclopedia.pub/entry/41939 (accessed July 28, 2026).
Pavan, M., & Moro, S. (2023, March 07). Tackling Pandemics through Computer-Aided Drug Discovery Approaches. In Encyclopedia. https://encyclopedia.pub/entry/41939
Pavan, Matteo and Stefano Moro. "Tackling Pandemics through Computer-Aided Drug Discovery Approaches." Encyclopedia. Web. 07 March, 2023.
Copy Citation
Since its outbreak in December 2019, the COVID-19 pandemic has caused the death of more than 6.5 million people around the world. The high transmissibility of its causative agent, the SARS-CoV-2 virus, coupled with its potentially lethal outcome, provoked a profound global economic and social crisis. The urgency of finding suitable pharmacological tools to tame the pandemic shed light on the ever-increasing importance of computer simulations in rationalizing and speeding up the design of new drugs, further stressing the need for developing quick and reliable methods to identify novel active molecules and characterize their mechanism of action.
COVID-19
SARS-CoV-2
rational drug design
CADD
SBDD
homology modeling
docking
pharmacophore
protein–ligand interaction fingerprints
molecular dynamics
1. Rational Design of COVID-19 Drugs
Several characteristics of the viral proteases family, including SARS-CoV-2 Mpro, make them an attractive target for the rational development of tailored drugs against COVID-19. First, the low sequence identity with human proteases coupled with distinct cleavage-site specificities reduces the possibility of off-target/side effects associated with the therapy [1]. Second, the striking conservation of protein fold and structural organization of the active site among different members of the same family leads to the possibility of developing pan-coronaviral drugs [2]. Third, the abundance of structural data about the SARS-CoV-2 main protease (659 structures have been deposited in the Protein Data Bank [3] to date) makes it possible to exploit the state-of-the-art structure-based approaches in drug design [4]. Furthermore, a similar strategy has already proved successful in finding efficient treatments against the hepatitis C virus [5][6] and human immunodeficiency virus (HIV) [7][8]. Finally, the experience acquired studying the original SARS-CoV protease [9], in conjunction with the rapid release to the scientific community of the SARS-CoV-2 protease [10], certainly played a major role in determining its prominent place within most COVID-19 drug discovery campaigns. A detailed report on structural features of the 3CLpro protease that can guide the design of novel inhibitors can be found in the work of Xiong et al. [11].
The first attempts at finding SARS-CoV-2 Mpro inhibitors involved the repurposing of existing protease inhibitors. Particularly, the hepatitis C protease inhibitor Boceprevir [12][13] and the feline coronavirus 3CLpro inhibitor GC373 (derived from its prodrug GC376) [14] were found to be active in the low µM potency range against Mpro [15], with the latter being particularly interesting due its promiscuous anticoronaviral activity [16]. Both candidate drugs share a similar peptidomimetic scaffold, which entails the most prominent interaction features of the first identified ones [10].
Although these primary hit compounds present a good binding pattern, their evolution towards clinical candidates and drugs is prevented by two main factors: first, covalent inhibitors are usually associated with selectivity problems, due to their ability to react promiscuously with a plethora of nucleophile moieties [17]; second, the peptidomimetic scaffold is usually associated with suboptimal pharmacokinetic properties that affect the preferred route of administration [18].
In this regard, a step forward was obtained when the first SARS-CoV-2 Mpro inhibitors were able to reach clinical stage experimentation, namely PF-07304814 (lately renamed as Lufotrelvir), a prodrug for the active principle PF-00835231, and PF-07321332 (Nirmatrelvir).
Lufotrelvir was originally developed by Pfizer in 2002–2003 for the SARS-CoV virus and later repurposed against the SARS-CoV-2 due to the high similarities between the two proteases [19]. Due to its efficacy against several viral strains in preclinical studies [20][21], it was advanced to the clinical stages of experimentation, albeit quickly overcome by Nirmatrelvir thanks to its more favorable pharmacokinetic profile [22].
Contrary to Lufotrelvir, which, similar to Remdesivir, requires parenteral administration, Nirmatrelvir can be administered orally [23], a must-have characteristic for the widespread adoption of drugs [24][25]. Designed by Pfizer amid the pandemic through the rational modification of Lufotrelvir [26], the structure of Nirmatrelvir was officially presented to the general audience on 6 April at the American Chemical Society Spring 2021 meeting [27], only one year after the official start of its development process [26] (Figure 1).
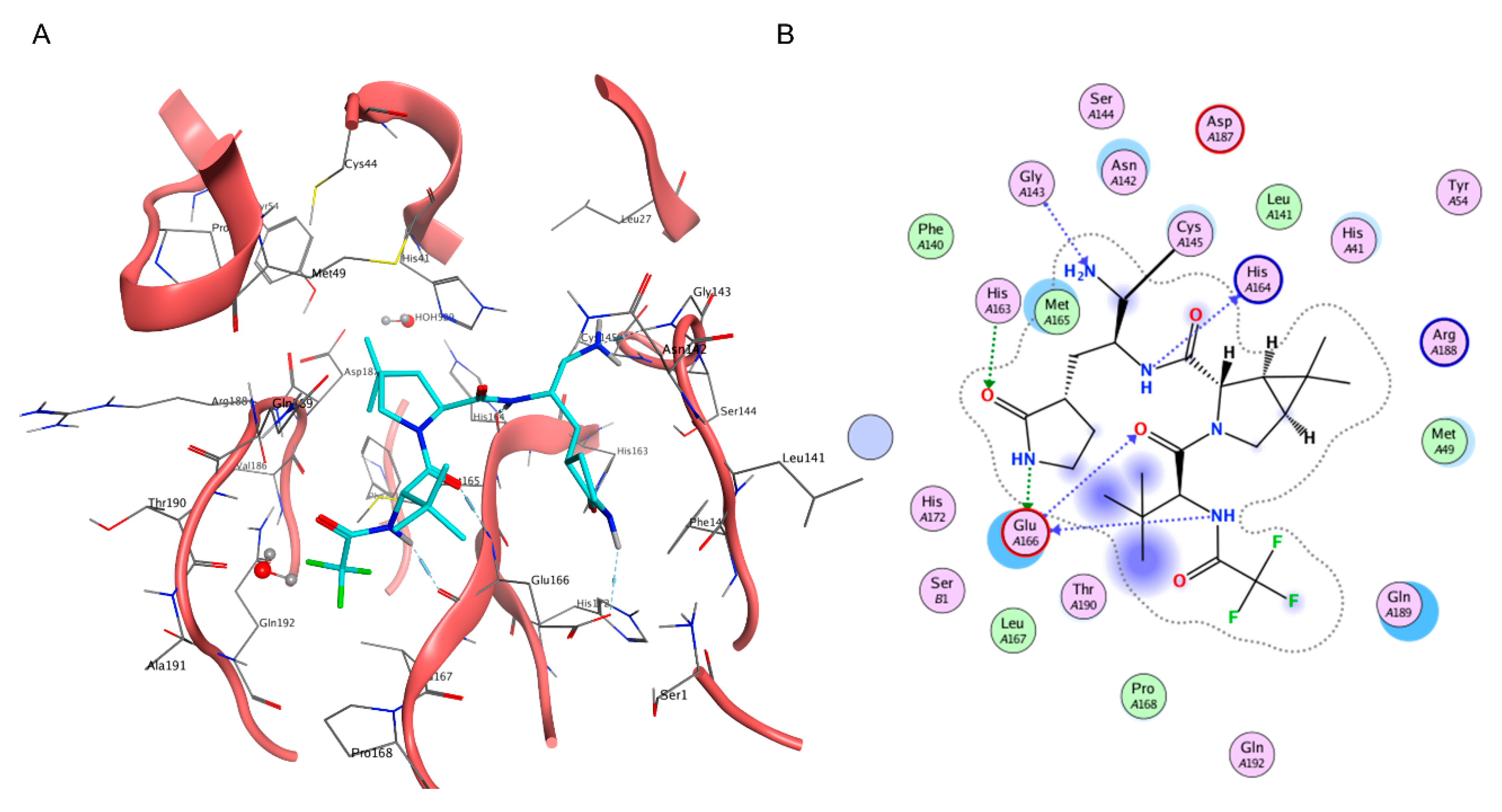
Figure 1. (A) Three-dimensional depiction of Nirmatrelvir orientation within the catalytic site of SARS-CoV-2 Mpro (PDB ID: 7RFW). (B) Bidimensional representation of intermolecular interactions of Nirmatrelvir–SARS-CoV-2 Mpro 7RFW complex.
This peptidomimetic inhibitor, which is administered in association with the pharmacokinetic enhancer Ritonavir and sold under the commercial name of Paxlovid, represents a hallmark in the history of both the COVID-19 pandemic and structure-based drug discovery, due to the groundbreaking speed of its discovery campaign [28]. Although clinical studies highlighted the remarkable therapeutic efficacy of Paxlovid in preventing the most severe COVID-19 cases [29], its effectiveness on more mild infections remains unclear [30]. Furthermore, the impact of viral mutations on present and future protease inhibitors has yet to be disclosed [31][32], thus justifying the current effort to find novel and diverse drugs that can enlarge the pool of pharmacological tools available against COVID-19.
An important step in this direction is represented by the development of Ensitrelvir (formerly known as S-217622), the first noncovalent, non peptidomimetic, orally available Mpro inhibitor to reach clinical stage experimentation [33]. This compound has successfully reached the third and final stage of clinical experimentation, thanks to its proven efficacy against mild-to-moderate or even asymptomatic infections [34][35]. Possible approval of this active principle by regulatory agencies would provide an additional and orthogonal therapeutic tool to Nirmatrelvir in the treatment of COVID-19 cases, thus reducing the impact of resistance mechanisms associated with the emergence of mutated viral strains [31][32].
2. Computer Simulations for Rational Drug Design
For most of its existence, the human genre has exploited natural products such as leaves, seeds, roots, bark, and flowers as medicines, based on empirical observations purely based on symptom relief [36][37].
Nevertheless, throughout the latest two centuries, the process of drug discovery has evolved rapidly from the serendipitous discovery of novel active principles derived from or inspired by natural compounds [38][39] to the rational design of brand-new chemical entities [40].
The major turning point in the history of modern drug discovery can be traced back to the 1980s when experimentally solved macromolecular structures become routinely available [41]. The enhanced accessibility of structural data about biological targets is reflected in a rapid interest in the development of computational methods that could valorize this information and aid medicinal chemists’ work [42].
Today, computer simulations are a staple point of drug discovery campaigns, thanks to their ability to streamline and reduce their attrition rate [43]. From a functional perspective, computer-aided drug discovery (CADD) techniques are employed in the earliest stages of the pipeline for hit identification, hit-to-lead optimization, and pharmacokinetic evaluations [44].
CADD methodologies can either fall into one of two subgroups, based on the rationale behind them: the first group is represented by ligand-based (LBDD) approaches, while the second one includes structure-based (SBDD) methods [45]. The main difference between these two orthogonal and complementary approaches is that the first one does not exploit any information about the target macromolecule structure (e.g., a protein or a nucleic acid), while the second one does [46].
Nowadays, with the advent of cryo-electron microscopy (cryo-EM) [47] and groundbreaking tools for de novo prediction of protein structures such as AlphaFold [48], the second approach has become the gold standard [4].
2.1. CADD Strategies against COVID-19
The starting point of every SBDD campaign is the identification of a target macromolecule (a protein or a nucleic acid) that is involved in the etiology and or pathogenesis of a disease of interest, whose function can be opportunely modulated through a specifically designed ligand, usually a small organic molecule [4].
Once the target has been identified, its structure must be retrieved, either through experimental methods such as X-Ray crystallography (XRC, the gold standard) [49], nuclear magnetic resonance (NMR) [50], and cryo-EM [51] or hypothesized through homology modeling or de novo prediction [52].
Homology modeling involves using a homologous protein with a high primary sequence identity with the target as a template for constructing its three-dimensional model [53][54]. De novo prediction, instead, does not rely on any information about other proteins’ structures and outputs a structural hypothesis that is solely based on the primary sequence of the target of interest [55].
While the second approach has gained a lot of momentum during the last two years, thanks to its unprecedentedly high accuracy [56][57], the first one is still relevant in those cases where important structural rearrangements occur between different states of the target functional cycle, other than predicting ligand-bound conformations [58][59].
In the context of the COVID-19 pandemic, where the extraordinary effort promoted by the scientific community quickly made several experimentally determined structures available, the relevance of structural modeling was highlighted by the ability to keep up with the high mutation rate of the virus [60][61], other than providing a useful starting point for drug discovery campaigns for a target whose structure had yet to be elucidated [62][63]. For example, several studies were conducted to investigate the impact of mutations found in both the spike protein [60][64][65][66][67][68] and the main protease [32][60][69][70] of emerging strains on viral fitness and resistance to existing therapies. These studies showed that relatively inexpensive approaches such as homology modeling and positional scanning could be reliable tools to rationalize the origin of the virus [69][71][72][73], quickly track the evolution of the original strain [60][74][75], predict the impact of future possible mutations [65][67] and adjust existing therapeutics tools accordingly [32][76].
The huge amount of structural information available on several SARS-CoV-2 druggable targets was fertile terrain for various COVID-19 SBDD campaigns [77][78], both in academia and in industry, with the most effort aimed at hitting well-characterized and pivotal viral targets such as Mpro or spike [79][80].
A remarkable example is represented by the COVID Moonshot Consortium, a drug discovery campaign driven by a collaborative effort among different research groups across the world aimed at targeting the SARS-CoV-2 main protease. This project led to the advancement of novel noncovalent orally available nanomolar Mpro inhibitors to clinical stage experimentation [81].
2.2. The Swiss Knife of SBDD: Molecular Docking
Within every SBDD campaign, available information about the target structure is exploited to fetch molecules able to recognize it selectively and potently [82]. Usually, this involves the identification of molecules that have good steric and electrostatic complementarity with the active site [83]. Depending on the steric and volumetric features of the binding site, the ligand type can be chosen accordingly, with small organic molecules being a better solution for buried cavities [84] and peptides, aptamers, or antibodies a better one for larger, flatter, and solvent-exposed interaction surfaces [85].
To narrow down the list of potentially active molecules to experimentally test to a feasible number, and to avoid wasting resources on compounds that do not possess the appropriate features to interact with the target, most SBDD campaigns start with a virtual screening process (SBVS) [86]. The most widely and successfully adopted method for SBVS is molecular docking, a computational protocol developed in the 1980s by Kuntz et al. [87] for predicting the preferred orientation of a certain ligand within the active site of a receptor [88].
Each docking program has two major components, which cooperate to find the solution to the protein–ligand docking problem [89]. The first part is the search algorithm (SA), which explores the ligand degrees of freedom within a user-defined search space centered around the active site of the protein [90]. The SA generates several ligand conformations (poses) that are fed to the second element of the program, i.e., the scoring function (SF), which qualitatively evaluates subsisting protein–ligand interaction features [91].
In the context of the COVID-19 pandemic, docking was also the king of computational methods used for drug discovery, thanks to the combination of its accuracy [92] and rapidity, which allows it to virtually screen billions of compounds in just a few days [93][94][95].
For example, Corona et al. reported the discovery of four low micromolar nsp13 inhibitors through a virtual screening carried out with the LiGen [96] docking program on an in-house natural compounds library [97].
Kolarič et al. identified two micromolar SARS-CoV-2 cell-entry inhibitors that act by binding human neuropilin-1 (nrp-1) and preventing its interaction with the spike protein, by performing a virtual screening with the GOLD [98] program on a library of commercially available compounds [99].
Vatansever et al. performed a virtual screening based on the Autodock [100] program on a library of drugs approved by the Food and Drug Administration and by the European Medical Agency (EMA) to discover six micromolar Mpro inhibitors [101].
Kao et al. reported the discovery of three sub-micromolar, synergistic nsp1 inhibitors identified through two independently executed virtual screenings with ICM [102][103] and Vina [104] software on a library of FDA-approved drugs [105].
Zhang et al. identified 11 natural compound Mpro inhibitors active in the low micromolar range through a virtual screening purely based on the commercial software Glide [106], developed by Schrödinger [107]. Another strategic use of docking-based virtual screening based on the Glide program is portrayed by the work of Huff et al., which designed six mixed covalent and noncovalent nanomolar Mpro inhibitors [108]. Another Glide-based virtual screening performed by Liu et al. led to the repurposing of histone deacetylase (HDAC) inhibitors as SARS-CoV-2 cell entry inhibitors through allosteric modulation of ACE2 and alteration of its ability to recognize the spike protein [109].
Wang et al. used LibDock [110] to perform a virtual screening on a library composed of FDA-approved peptides. This led to the identification of a nanomolar SARS-CoV-2 cell entry inhibitor that exerts its effect by binding to the human ACE2 receptor [111].
A remarkable result was obtained by Luttens et al., which identified eight Mpro inhibitors (including a nanomolar compound with pan coronaviral activity) by combining fragment-based drug design with ultra-large virtual screening based on the DOCK [87] program [112].
Welker et al. exploited the molecular docking pipeline of the LeadIT [113] program to repurpose previously identified SARS-CoV PLpro inhibitors towards its SARS-CoV-2 homolog, demonstrating their activity on viral replication in cell-based assays [114].
Otava et al. utilized docking calculations with the GOLD [98] software to rationalize the structure–activity relationship of a series of rationally designed S-adenosyl-L-homocysteine derivatives, some of which showed inhibitory activity towards SARS-CoV-2 nsp14 in the low nanomolar potency range [115].
Similarly, Wang et al. exploited docking with Vina to rationalize the SAR of a series of rationally designed phenanthridine nucleocapsid protein (NPro) inhibitors, including two compounds showing low micromolar inhibitory activity [116].
2.3. Complementary Strategies to Address Docking Limitations
Although a very efficient and useful tool, molecular docking is rarely used on its own within SBDD campaigns and, indeed, is most often coupled with other methods to compensate for its weak points, such as neglecting receptor flexibility or the role of solvents [117], thus increasing the virtual screening success rate [118]. Another major limitation is represented by the poor ranking capabilities of classical scoring functions [119], which is the main cause of the high false positive rate of docking-based virtual screenings [120]. Indeed, in order to be universally applicable across different biological targets and computationally efficient enough to evaluate a large number of compounds, scoring functions have some limitations in the physical description of the binding event, which prevent any correlation between docking scores and experimentally determined affinity values [91]. Furthermore, little to no difference in score exists between top-ranking compounds derived from large virtual screening campaigns, making it practically impossible to distinguish active from inactive compounds solely based on the docking score [121]. For these reasons, each docking-based virtual screening cannot be blindly executed and fully automatized, and a careful setup of the experiment must be executed based on the available literature data and the knowledge of the target [121][122]. For COVID-19, the importance of this common-sense medicinal chemistry practice has been highlighted by the retrospective literature analysis provided by Llanos et al., which showcased the poor performances of structure-based virtual screenings solely based on ranking provided by docking scoring functions [118].
A possible solution to the limited physical description of the protein–ligand binding event of docking is to couple it with molecular dynamics (MD) simulations [89][123]. Molecular dynamics is a computational technique that allows investigating the time-dependent evolution of biological systems following the rules of molecular mechanics, i.e., determining the atomic trajectories by numerically solving Newton’s equation of motion, where forces between the particles and their potential energies are calculated according to molecular mechanical force fields [124]. Due to the heavy computational workload required to run these types of simulations, MD is rarely used for screening purposes, while it is more frequently exploited for the refinement of docking results, i.e., evaluating the pose stability or optimizing the protein–ligand complex geometry for a more accurate estimation of the free binding energy [125][126].
Regarding the pitfalls of the scoring component of docking programs, one possible strategy is to apply some form of knowledge-based filter upon docking results, in a similar fashion to what would happen if each pose were visually inspected [127]. For example, experimental information about critical protein–ligand interactions required for binding can be encoded within a pharmacophore filter or an interaction fingerprint, both of which can be used as constraints in the pose selection process [128]. In the case of pharmacophore filters, poses are filtered based on their ability to place a given functional group within a defined volume [129][130], while in the case of protein–ligand interaction fingerprint, the selection is usually based on the similarity between the reference and the query vector, representing the interaction features of the reference compound (a true active) and the investigated molecule respectively [131][132].
For instance, Wang et al. used a combination of structure-based pharmacophore screening, docking (both performed with the appropriate tools of the Molecular Operating Environment suite), and post-docking molecular dynamics refinement to identify a set of four sub-micromolar Mpro inhibitors among a database of in-house compounds [133].
The same protocol was successfully exploited by Tian et al. to identify four sub-micromolar PLpro inhibitors in the same in-house library [134].
Furthermore, a slight variation of the protocol was also employed by Yin et al. to discover a noncovalent cyclic peptide that simultaneously inhibits both SARS-CoV-2 Mpro and nrp-1 with an activity in the low nanomolar range [135]. Within this scientific work, pharmacophore constraints were used for scoring peptide poses on Mpro, while traditional docking scores were used for the nrp-1 screening.
A remarkable joint computational work by Gossen et al. led to the molecular dynamics-driven design of a structure-based pharmacophore filter, which was then exploited to identify two nanomolar Mpro inhibitors among a library of publicly available compounds [136].
A similar approach was exploited by Hu et al., which exploited the combination between MD-based pharmacophore filtering, docking-based virtual screening within the Molecular Operating Environment suite, and MD-based post-docking refinement to identify micromolar SARS-CoV-2 cell entry inhibitors targeting the FP of the spike protein [137].
Jang et al. used protein–ligand interaction fingerprint similarity as a post-docking filter for their double virtual screening on both Mpro and RdRp with the Vina program to identify seven compounds inhibiting SARS-CoV-2 replication in cell-based assays among a library of approved drugs [138].
Due to the static nature of molecular docking, which does not consider receptor flexibility, the choice of the input structure is vital for the success rate of a virtual screening [139]. Although molecular dynamics can be a useful posterior refinement of poses, a wrong input conformation of the target macromolecule could prevent the sampling of native-like poses for active compounds, leading to a reduced hit-finding rate [140]. For this reason, multiple conformations of the same receptor derived from MD simulations or experimentally solved in different conditions can be used in parallel in a process defined as ensemble docking (ED) [141]. When this approach is used, docking calculations are independently run on each structure, with virtual hit compounds being identified either through consensus scoring or a consensus ranking approach [142][143]. In the case of consensus scoring, the docking score of the same molecule is averaged across the different virtual screenings, with the final ranking based on the consensus score [144]. Differently, consensus ranking involves the selection of top-ranking hit compounds across different virtual screenings, regardless of congruence between scores [145]. A consensus approach can also be utilized to rank molecules based on virtual screening executed on the same receptor structures with different docking protocols [146].
For example, Gimeno et al. applied a consensus scoring approach to three independently executed virtual screenings through Glide, FRED [147], and Vina software to identify two Mpro micromolar inhibitors within the Drugbank database, a library that includes all drugs approved by the Food and Drug Administration (FDA) [148].
Yang et al., instead, employed an ensemble docking approach with the Glide docking software to identify six Mpro inhibitors among a library of commercially available peptidomimetic compounds, two of which demonstrated sub-micromolar potency [149].
Rubio-Martinez et al. used a combination of ensemble docking based on QVina2 [150] and post-docking molecular dynamics refinement to identify five Mpro micromolar inhibitors within a library of commercially available natural compounds [151].
A mixture of the previous two approaches was exploited by Clyde et al. for their High-Throughput Virtual Screening (HTVS), based on both ensemble docking and consensus scoring between the FRED and Vina docking programs, that led to the discovery of seven micromolar Mpro inhibitors among a set of commercially available compounds [152].
Further, a combination of consensus ranking among Autodock, Hybrid, and FlexX and post-docking molecular dynamics refinement was utilized by Glaab et al. to virtually screen a library of commercially available compounds and identify two micromolar Mpro inhibitors [153].
Similarly, Ghahremanpour et al. applied both consensus ranking among three independent virtual screenings performed with the Glide, Autodock, and Vina software and post-docking molecular dynamics refinement to identify 14 micromolar Mpro inhibitors within the Drugbank database [154].
Another possible solution to cope with inaccuracy in free binding energy determination by traditional scoring functions is to rescore docking poses using more computationally intensive and accurate methods such as Free Energy Perturbation (FEP) [155] or MMGBSA/MMPBSA [156]. The first approach relies on performing a series of alchemical transformations across a set of ligands that need to be evaluated. This conversion cycle allows calculating relative differences in the free binding energy that can be used for a more accurate ranking of hit compounds derived from a virtual screening [157]. The second approach relies instead on correcting the gas phase interaction energy calculated according to the molecular mechanics force field with a term accounting for the desolvation-free energy, where the polar component is estimated either by numerically solving the Poisson–Boltzmann equation (MMPBSA) or through the Generalized Born method (MMGBSA) [158].
Intriguingly, one of the hit compounds identified in the work of Ghahremanpour et al. was then used by Zhang et al. for the FEP-driven design of multiple nanomolar Mpro inhibitors [159].
A similar combination of Glide docking and FEP to determine the absolute binding free energy was also employed by Li et al. to identify 15 micromolar Mpro inhibitors within the Drugbank database [160]. The efficacy of FEP in estimating the binding energy of potential Mpro inhibitors was also highlighted by a retrospective study by Ngo et al. [161].
A multistep virtual screening involving semiflexible docking with Glide, Schrödinger induced-fit docking [162], MD-based post-docking refinement, and binding free energy estimation with the MMGBSA [163] protocol was exploited by Ibrahim et al. to identify one low micromolar nsp15 inhibitor [164].
Although the estimation of thermodynamic properties such as the free binding energy has been a staple point of drug discovery campaigns, both from a computational and an experimental perspective, lately there has been a major interest shift towards the determination of kinetic parameters since they better correlate with in vivo efficacy [165]. Specifically, several MD-based methods have been developed throughout the years to rank compounds based on their predicted residence time, i.e., the time that the ligand spends in the receptor-bound state [166]. Among those, Pavan et al. developed Thermal Titration Molecular Dynamics (TTMD), a new method for qualitative estimation of protein–ligand complex stability (Figure 2), which was successfully applied for correctly discriminating tight, low nanomolar binders from weak, micromolar SARS-CoV-2 Mpro inhibitors [167].
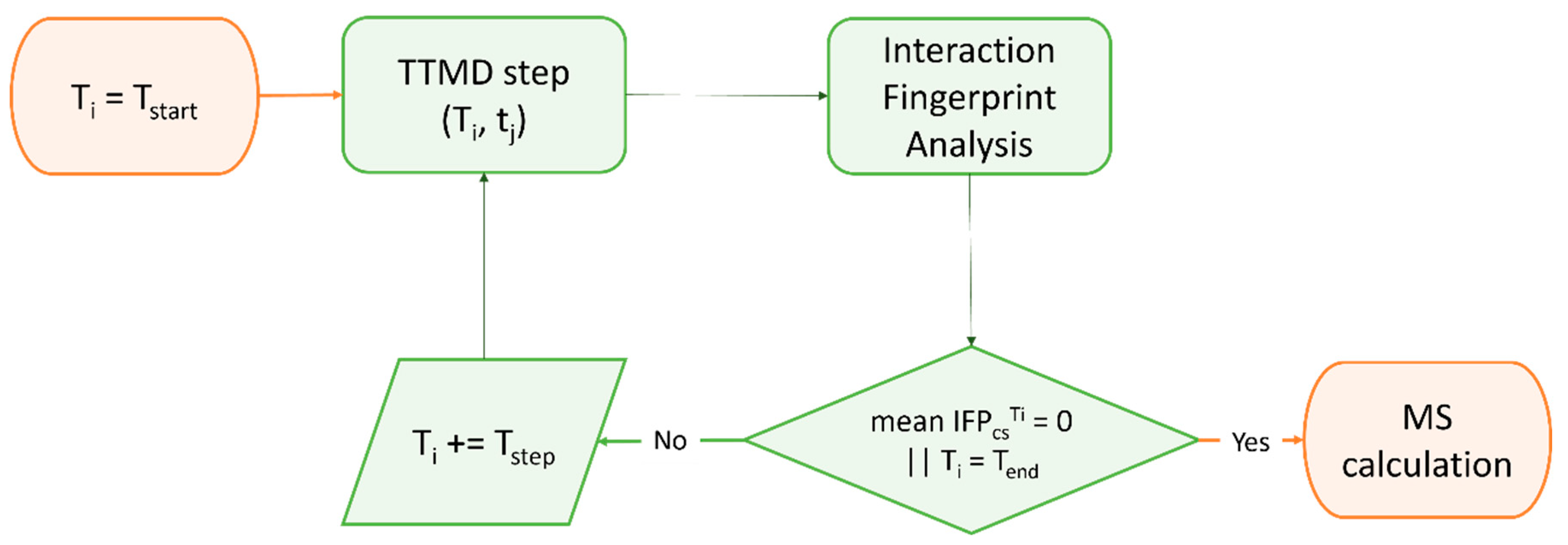
Figure 2. Workflow of a Thermal Titration Molecular Dynamics (TTMD) simulation. The time-dependent conservation of the native binding mode within a protein–ligand complex of interest is monitored with a scoring function based on interaction fingerprint through a series of short molecular dynamics simulations performed at progressively increasing temperatures. The simulation is carried out until the target temperature is reached or the dissociation process is completed. A coefficient called MS is then calculated and used to rank ligands based on the persistence of their native binding mode.
2.4. Beyond Protein-Ligand Docking: Alternative Strategies for Rational Drug Development
Despite the indisputable relevance of molecular docking within most SARS-CoV-2 drug discovery campaigns, other approaches were successfully implemented, especially for projects which deviate from the design of a standard small molecule noncovalent binder.
For example, Zaidman et al. developed Covalentizer, an automated pipeline for the conversion of noncovalent binders to irreversible ones, which was successfully applied to the conversion of a SARS-CoV Mpro reversible inhibitor to a sub-micromolar SARS-CoV-2 Mpro irreversible one [168].
Valiente et al. reported the discovery of D-peptides that bind the spike RBD with low nanomolar affinity, hence blocking SARS-CoV-2 infection in cell-based assays. These ACE2-mimicking peptides were selected within the starting library through a combination of structural alignment, MD-based post-docking refinement, and binding free energy estimation [169].
Similarly, a series of peptides mimicking the HR2 domain of the spike protein able to prevent SARS-CoV-2 infection in cell-based assays with low micromolar potency were designed through the combination between structural alignment, mutational scanning with the BeAtMuSiC [170] tool, and MD-based post-docking refinement [171].
Jeong et al. used Rosetta [172] to rationally design a mAb that recognizes a conserved surface on the spike RBD of various coronaviruses with picomolar binding affinities, thereby strongly inhibiting SARS-CoV-2 replication in cell-based assay [173].
A similar strategy was exploited by Miao et al., which employed Rosetta docking and MD-based post-docking refinement to design an RNA aptamer that binds with picomolar affinity to the spike RBD and inhibits SARS-CoV-2 replication with sub-micromolar potency in cell-based assay [174].
Further, Cao et al. utilized a combination of modeling with Rosetta and docking with RifDock [175] to design ten mini proteins which bind with picomolar affinity to the spike RBD thus inhibiting SARS-CoV-2 infection within cell-based assays [176].
Moreover, Glasgow et al. combined modeling with Rosetta and computational alanine scanning with Robetta [177][178] to rationally design “ACE2 receptor traps”, i.e., engineered proteins that bind the spike RBD with high affinity and neutralize SARS-CoV-2 infection as effectively as clinically used mAbs [179].
As thoroughly discussed in previous paragraphs, many SARS-CoV-2 drug discovery campaigns favored static, time-independent approaches such as docking or structural alignment, over time-dependent methods such as molecular dynamics. This can be attributed to the long calculation times, the reduced conformational sampling capabilities, and the lower accessibility of MD simulations to the general medicinal chemistry audience [126][180]. Despite these issues, several works demonstrated the potential of using full-fledged MD-based drug discovery pipelines, especially when smart enhanced-sampling strategies are employed [180].
For example, Bissaro et al. showed how high-throughput supervised molecular dynamics (HT-SuMD) [181], a virtual screening platform based on an enhanced sampling MD protocol, could be successfully exploited for docking fragments to the active site of SARS-CoV-2 Mpro, overcoming accuracy limitations of most docking protocols [182] in identifying the native-like binding mode for frag-like compounds [183].
Furthermore, the SuMD [184][185] algorithm (Figure 3) was successfully exploited by Pavan et al. to decipher details about the recognition mechanism of Nirmatrelvir upon the SARS-CoV-2 Mpro catalytic site before any structural detail was revealed by the drug developer, with successive structural [23] and molecular medicine [32] studies confirming the prediction validity [186].
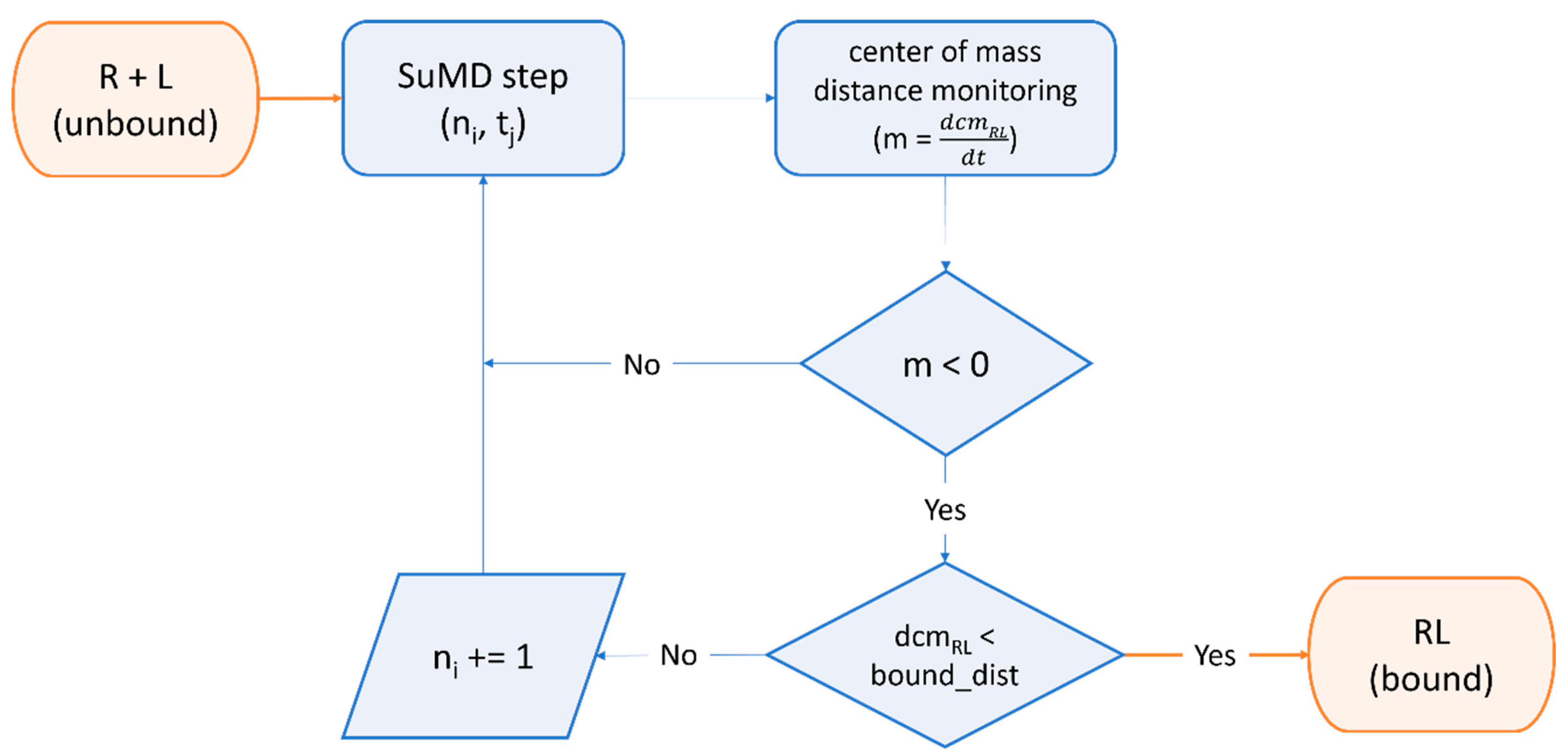
Figure 3. Workflow of a Supervised Molecular Dynamics (SuMD) simulation. The ligand is dynamically docked within a user-defined binding site through a series of short, unbiased molecular dynamics simulations. At the end of each step, the distance of mass between the ligand and the receptor binding site is computed for each trajectory frame and is fed to a tabu-like algorithm. If the slope of the straight line that interpolates the data is negative, indicating the ligand is approaching the binding site, the step is retained, and the simulation continues with the next “SuMD-step”. If not, the step is discarded and repeated, randomly reassigning particles’ velocities through the Langevin thermostat. This cycle is repeated until a threshold distance is reached or other user-defined termination criteria are met.
Moreover, an evolved version of the SuMD protocol was developed by Pavan et al. and successfully applied to the study of the recognition mechanism between RNA aptamers and proteins, including an RNA-aptamer that binds to the spike RBD with picomolar affinity thus preventing the viral infection of host cells [187].
References
- de Clercq, E. Strategies in the Design of Antiviral Drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25.
- Kilianski, A.; Baker, S.C. Cell-Based Antiviral Screening against Coronaviruses: Developing Virus-Specific and Broad-Spectrum Inhibitors. Antiviral Res. 2014, 101, 105–112.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242.
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797.
- Njoroge, F.G.; Chen, K.X.; Shih, N.Y.; Piwinski, J.J. Challenges in Modern Drug Discovery: A Case Study of Boceprevir, an HCV Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. Acc. Chem. Res. 2008, 41, 50–59.
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From Non-A, Non-B Hepatitis to Hepatitis C Virus Cure. J. Hepatol. 2015, 62, S87–S99.
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 Protease: A Major Success of Structure-Assisted Drug Design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284.
- Palella, F.J.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998, 338, 853–860.
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.E.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small Molecules Targeting Severe Acute Respiratory Syndrome Human Coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017.
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293.
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What Coronavirus 3C-like Protease Tells Us: From Structure, Substrate Selectivity, to Inhibitor Design. Med. Res. Rev. 2021, 41, 1965–1998.
- Poordad, F.; McCone, J.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for Untreated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1195–1206.
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1207–1217.
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like Protease Inhibitor in Treating Various Forms of acquired Feline Infectious Peritonitis. J. Feline Med. Surg. 2018, 20, 378.
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692.
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282.
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628.
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747.
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical Characterization of an Intravenous Coronavirus 3CL Protease Inhibitor for the Potential Treatment of COVID19. Nat. Commun. 2021, 12, 6055.
- De Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A Comparative Analysis of SARS-CoV-2 Antivirals Characterizes 3CLpro Inhibitor PF-00835231 as a Potential New Treatment for COVID-19. J. Virol. 2021, 95, e01819-20.
- Vandyck, K.; Deval, J. Considerations for the Discovery and Development of 3-Chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 2021, 49, 36.
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 M pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26.
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341.
- How Pfizer Scientists Transformed an Old Drug Lead into a COVID-19 Antiviral. Available online: https://cen.acs.org/pharmaceuticals/drug-discovery/How-Pfizer-scientists-transformed-an-old-drug-lead-into-a-COVID-19-antiviral/100/i3 (accessed on 14 December 2022).
- Pfizer Unveils Its Oral SARS-CoV-2 Inhibitor. Available online: https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13 (accessed on 14 December 2022).
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591.
- Mahase, E. COVID-19: Pfizer’s Paxlovid Is 89% Effective in Patients at Risk of Serious Illness, Company Reports. BMJ 2021, 375, n2713.
- Hung, Y.P.; Lee, J.C.; Chiu, C.W.; Lee, C.C.; Tsai, P.J.; Hsu, I.L.; Ko, W.C. Oral Nirmatrelvir/Ritonavir Therapy for COVID-19: The Dawn in the Dark? Antibiotics 2022, 11, 220.
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main Protease Mutants of SARS-CoV-2 Variants Remain Susceptible to Nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629.
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D.; et al. SARS-CoV-2 3CLpro Mutations Selected in a VSV-Based System Confer Resistance to Nirmatrelvir, Ensitrelvir, and GC376. Sci. Transl. Med. 2022, 15, eabq7360.
- McCarthy, M.W. Ensitrelvir as a Potential Treatment for COVID-19. Expert Opin. Pharmacother. 2022, 23, 1995–1998.
- Shimizu, R.; Sonoyama, T.; Fukuhara, T.; Kuwata, A.; Matsuo, Y.; Kubota, R. Safety, Tolerability, and Pharmacokinetics of the Novel Antiviral Agent Ensitrelvir Fumaric Acid, a SARS-CoV-2 3CL Protease Inhibitor, in Healthy Adults. Antimicrob. Agents Chemother. 2022, 66, e00632-22.
- Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; et al. A Randomized Phase 2/3 Study of Ensitrelvir, a Novel Oral SARS-CoV-2 3C-Like Protease Inhibitor, in Japanese Patients with Mild-to-Moderate COVID-19 or Asymptomatic SARS-CoV-2 Infection: Results of the Phase 2a Part. Antimicrob. Agents Chemother. 2022, 66, e00697-22.
- Cragg, G.M.; Newman, D.J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695.
- Gurib-Fakim, A. Medicinal Plants: Traditions of Yesterday and Drugs of Tomorrow. Mol. Asp. Med. 2006, 27, 1–93.
- Harvey, A.L. Natural Products in Drug Discovery. Drug Discov. Today 2008, 13, 894–901.
- Ban, T.A. The Role of Serendipity in Drug Discovery. Dialogues Clin. Neurosci. 2006, 8, 335–344.
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651.
- Jaskolski, M.; Dauter, Z.; Wlodawer, A. A Brief History of Macromolecular Crystallography, Illustrated by a Family Tree and Its Nobel Fruits. FEBS J. 2014, 281, 3985–4009.
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395.
- Leelananda, S.P.; Lindert, S. Computational Methods in Drug Discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718.
- Kapetanovic, I.M. Computer-Aided Drug Discovery and Development (CADDD): In Silico-Chemico-Biological Approach. Chem. Biol. Interact. 2008, 171, 165–176.
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharm. Res. 2015, 38, 1686–1701.
- Yu, W.; Mackerell, A.D. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106.
- Bai, X.C.; McMullan, G.; Scheres, S.H.W. How Cryo-EM Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589.
- Smyth, M.S.; Martin, J.H.J. X Ray Crystallography. Mol. Pathol. 2000, 53, 8.
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, e1000168.
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) Is the Limit. Int. J. Mol. Sci. 2019, 20, 4186.
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697.
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325.
- Cavasotto, C.N.; Phatak, S.S. Homology Modeling in Drug Discovery: Current Trends and Applications. Drug Discov. Today 2009, 14, 676–683.
- Bonneau, R.; Strauss, C.E.M.; Rohl, C.A.; Chivian, D.; Bradley, P.; Malmströ, L.; Robertson, T.; Baker, D. De Novo Prediction of Three-Dimensional Structures for Major Protein Families. J. Mol. Biol. 2002, 322, 65–78.
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148.
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-Accuracy Protein Structure Prediction in CASP14. Proteins 2021, 89, 1687–1699.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1711–1721.
- Kwon, S.; Won, J.; Kryshtafovych, A.; Seok, C. Assessment of Protein Model Structure Accuracy Estimation in CASP14: Old and New Challenges. Proteins Struct. Funct. Bioinform. 2021, 89, 1940–1948.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. From the Wuhan-Hu-1 Strain to the XD and XE Variants: Is Targeting the SARS-CoV-2 Spike Protein Still a Pharmaceutically Relevant Option against COVID-19? J. Enzyme Inhib. Med. Chem. 2022, 37, 1704–1714.
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K.; et al. Evolution of the SARS-CoV-2 Proteome in Three Dimensions (3D) during the First 6 Months of the COVID-19 Pandemic. Proteins: Struct. Funct. Bioinform. 2022, 90, 1054–1080.
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.L.; Li, J. A Guideline for Homology Modeling of the Proteins from Newly Discovered Betacoronavirus, 2019 Novel Coronavirus (2019-NCoV). J. Med. Virol. 2020, 92, 1542–1548.
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788.
- Bassani, D.; Ragazzi, E.; Lapolla, A.; Sartore, G.; Moro, S. Omicron Variant of SARS-CoV-2 Virus: In Silico Evaluation of the Possible Impact on People Affected by Diabetes Mellitus. Front. Endocrinol. 2022, 13, 284.
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface Can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol. Biol. 2021, 433, 167051.
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601.e6.
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654.
- Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation Signatures and in Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Bat Coronaviruses Related to SARS-CoV-2: What about Their 3CL Proteases (MPro)? J. Enzym. Inhib. Med. Chem. 2022, 37, 1077–1082.
- Martin, R.W.; Butts, C.T.; Cross, T.J.; Takahashi, G.R.; Diessner, E.M.; Crosby, M.G.; Farahmand, V.; Zhuang, S. Sequence Characterization and Molecular Modeling of Clinically Relevant Variants of the SARS-CoV-2 Main Protease. Biochemistry 2020, 59, 3741–3756.
- Huang, X.; Zhang, C.; Pearce, R.; Omenn, G.S.; Zhang, Y. Identifying the Zoonotic Origin of SARS-CoV-2 by Modeling the Binding Affinity between the Spike Receptor-Binding Domain and Host ACE2. J. Proteome Res. 2020, 19, 4844–4856.
- Rodrigues, J.P.G.L.M.; Barrera-Vilarmau, S.; Teixeira, J.M.C.; Sorokina, M.; Seckel, E.; Kastritis, P.L.; Levitt, M. Insights on Cross-Species Transmission of SARS-CoV-2 from Structural Modeling. PLoS Comput. Biol. 2020, 16, e1008449.
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In Silico Comparison of SARS-CoV-2 Spike Protein-ACE2 Binding Affinities across Species and Implications for Virus Origin. Sci. Rep. 2021, 11, 13063.
- Sharma, P.; Kumar, M.; Tripathi, M.K.; Gupta, D.; Vishwakarma, P.; Das, U.; Kaur, P. Genomic and Structural Mechanistic Insight to Reveal the Differential Infectivity of Omicron and Other Variants of Concern. Comput. Biol. Med. 2022, 150, 106129.
- Jacob, J.J.; Vasudevan, K.; Pragasam, A.K.; Gunasekaran, K.; Veeraraghavan, B.; Mutreja, A. Evolutionary Tracking of SARS-CoV-2 Genetic Variants Highlights an Intricate Balance of Stabilizing and Destabilizing Mutations. mBio 2021, 12, e01188-21.
- Luo, R.; Delaunay-Moisan, A.; Timmis, K.; Danchin, A. SARS-CoV-2 Biology and Variants: Anticipation of Viral Evolution and What Needs to Be Done. Environ. Microbiol. 2021, 23, 2339–2363.
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and COVID-19 Therapeutics. ChemMedChem 2020, 15, 907–932.
- Ghosh, A.K.; Mishevich, J.L.; Mesecar, A.; Mitsuya, H. Recent Drug Development and Medicinal Chemistry Approaches for the Treatment of SARS-CoV-2 Infection and COVID-19. ChemMedChem 2022, 17, e202200440.
- Tiwari, V.; Beer, J.C.; Sankaranarayanan, N.V.; Swanson-Mungerson, M.; Desai, U.R. Discovering Small-Molecule Therapeutics against SARS-CoV-2. Drug Discov. Today 2020, 25, 1535–1544.
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral Drug Discovery: Preparing for the next Pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655.
- Consortium, T.C.M.; Achdout, H.; Aimon, A.; Bar-David, E.; Barr, H.; Ben-Shmuel, A.; Bennett, J.; Bilenko, V.A.; Bilenko, V.A.; Boby, M.L.; et al. Open Science Discovery of Oral Non-Covalent SARS-CoV-2 Main Protease Inhibitor Therapeutics. bioRxiv 2022.
- Kuntz, I.D. Structure-Based Strategies for Drug Design and Discovery. Science 1992, 257, 1078–1082.
- Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732–18758.
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389.
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of Protein Pockets and Cavities: Measurement of Binding Site Geometry and Implications for Ligand Design. Protein Sci. 1998, 7, 1884–1897.
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discov. 2004, 3, 935–949.
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. J. Mol. Biol. 1982, 161, 269–288.
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Current Computer Aided-Drug Design 2012, 7, 146–157.
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923.
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of Docking: An Overview of Search Algorithms and a Guide to Scoring Functions. Proteins Struct. Funct. Genet. 2002, 47, 409–443.
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931.
- Bassani, D.; Pavan, M.; Bolcato, G.; Sturlese, M.; Moro, S. Re-Exploring the Ability of Common Docking Programs to Correctly Reproduce the Binding Modes of Non-Covalent Inhibitors of SARS-CoV-2 Protease Mpro. Pharmaceuticals 2022, 15, 180.
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028.
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to COVID-19. J. Chem. Inf. Model. 2020, 60, 5832–5852.
- Gorgulla, C.; Padmanabha Das, K.M.; Leigh, K.E.; Cespugli, M.; Fischer, P.D.; Wang, Z.F.; Tesseyre, G.; Pandita, S.; Shnapir, A.; Calderaio, A.; et al. A Multi-Pronged Approach Targeting SARS-CoV-2 Proteins Using Ultra-Large Virtual Screening. iScience 2021, 24, 102021.
- Manelfi, C.; Gossen, J.; Gervasoni, S.; Talarico, C.; Albani, S.; Philipp, B.J.; Musiani, F.; Vistoli, G.; Rossetti, G.; Beccari, A.R.; et al. Combining Different Docking Engines and Consensus Strategies to Design and Validate Optimized Virtual Screening Protocols for the SARS-CoV-2 3CL Protease. Molecules 2021, 26, 797.
- Corona, A.; Wycisk, K.; Talarico, C.; Manelfi, C.; Milia, J.; Cannalire, R.; Esposito, F.; Gribbon, P.; Zaliani, A.; Iaconis, D.; et al. Natural Compounds Inhibit SARS-CoV-2 Nsp13 Unwinding and ATPase Enzyme Activities. ACS Pharmacol. Transl. Sci. 2022, 5, 226–239.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins 2003, 52, 609–623.
- Kolarič, A.; Jukič, M.; Bren, U. Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1. Pharmaceuticals 2022, 15, 165.
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785.
- Vatansever, E.C.; Yang, K.S.; Drelich, A.K.; Kratch, K.C.; Cho, C.C.; Kempaiah, K.R.; Hsu, J.C.; Mellott, D.M.; Xu, S.; Tseng, C.T.K.; et al. Bepridil Is Potent against SARS-CoV-2 in Vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2012201118.
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and Scoring with ICM: The Benchmarking Results and Strategies for Improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686.
- Lam, P.C.H.; Abagyan, R.; Totrov, M. Ligand-Biased Ensemble Receptor Docking (LigBEnD): A Hybrid Ligand/Receptor Structure-Based Approach. J. Comput. Aided Mol. Des. 2018, 32, 187–198.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455.
- Kao, H.T.; Orry, A.; Palfreyman, M.G.; Porton, B. Synergistic Interactions of Repurposed Drugs That Inhibit Nsp1, a Major Virulence Factor for COVID-19. Sci. Rep. 2022, 12, 10174.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749.
- Zhang, Y.; Gao, H.; Hu, X.; Wang, Q.; Zhong, F.; Zhou, X.; Lin, C.; Yang, Y.; Wei, J.; Du, W.; et al. Structure-Based Discovery and Structural Basis of a Novel Broad-Spectrum Natural Product against the Main Protease of Coronavirus. J. Virol. 2022, 96, 1253–1274.
- Huff, S.; Kummetha, I.R.; Tiwari, S.K.; Huante, M.B.; Clark, A.E.; Wang, S.; Bray, W.; Smith, D.; Carlin, A.F.; Endsley, M.; et al. Discovery and Mechanism of SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2866–2879.
- Liu, K.; Zou, R.; Cui, W.; Li, M.; Wang, X.; Dong, J.; Li, H.; Li, H.; Wang, P.; Shao, X.; et al. Clinical HDAC Inhibitors Are Effective Drugs to Prevent the Entry of SARS-CoV2. ACS Pharmacol. Transl. Sci. 2020, 3, 1361–1370.
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation Studies of the Site-Directed Docking Program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171.
- Wang, G.; Yang, M.L.; Duan, Z.L.; Liu, F.L.; Jin, L.; Long, C.B.; Zhang, M.; Tang, X.P.; Xu, L.; Li, Y.C.; et al. Dalbavancin Binds ACE2 to Block Its Interaction with SARS-CoV-2 Spike Protein and Is Effective in Inhibiting SARS-CoV-2 Infection in Animal Models. Cell Res. 2020, 31, 17–24.
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; de Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc 2022, 144, 2905–2920.
- Cross, S.S.J. Improved FlexX Docking Using FlexS-Determined Base Fragment Placement. J. Chem. Inf. Model. 2005, 45, 993–1001.
- Welker, A.; Kersten, C.; Müller, C.; Madhugiri, R.; Zimmer, C.; Müller, P.; Zimmermann, R.; Hammerschmidt, S.; Maus, H.; Ziebuhr, J.; et al. Structure-Activity Relationships of Benzamides and Isoindolines Designed as SARS-CoV Protease Inhibitors Effective against SARS-CoV-2. ChemMedChem 2021, 16, 340–354.
- Otava, T.; Šála, M.; Li, F.; Fanfrlík, J.; Devkota, K.; Perveen, S.; Chau, I.; Pakarian, P.; Hobza, P.; Vedadi, M.; et al. The Structure-Based Design of SARS-CoV-2 Nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors. ACS Infect Dis. 2021, 7, 2214–2220.
- Wang, Y.T.; Long, X.Y.; Ding, X.; Fan, S.R.; Cai, J.Y.; Yang, B.J.; Zhang, X.F.; Luo, R.H.; Yang, L.; Ruan, T.; et al. Novel Nucleocapsid Protein-Targeting Phenanthridine Inhibitors of SARS-CoV-2. Eur. J. Med. Chem. 2022, 227, 113966.
- Chen, Y.C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95.
- Llanos, M.A.; Gantner, M.E.; Rodriguez, S.; Alberca, L.N.; Bellera, C.L.; Talevi, A.; Gavernet, L. Strengths and Weaknesses of Docking Simulations in the SARS-CoV-2 Era: The Main Protease (Mpro) Case Study. J. Chem. Inf. Model. 2021, 61, 3758–3770.
- Chaput, L.; Mouawad, L. Efficient Conformational Sampling and Weak Scoring in Docking Programs? Strategy of the Wisdom of Crowds. J. Cheminform. 2017, 9, 37.
- Neves, B.J.; Mottin, M.; Moreira-Filho, J.T.; de Paula Sousa, B.K.; Mendonca, S.S.; Andrade, C.H. Best Practices for Docking-Based Virtual Screening. Mol. Docking Comput. Aided Drug Des. 2021, 2021, 75–98.
- Cerón-Carrasco, J.P. When Virtual Screening Yields Inactive Drugs: Dealing with False Theoretical Friends. ChemMedChem 2022, 17, e202200278.
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881.
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining Docking and Molecular Dynamic Simulations in Drug Design. Med. Res. Rev. 2006, 26, 531–568.
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129.
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652.
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 1–9.
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421.
- Tan, L.; Batista, J.; Bajorath, J. Computational Methodologies for Compound Database Searching That Utilize Experimental Protein-Ligand Interaction Information. Chem. Biol. Drug Des. 2010, 76, 191–200.
- Peach, M.L.; Nicklaus, M.C. Combining Docking with Pharmacophore Filtering for Improved Virtual Screening. J. Cheminform. 2009, 1, 6.
- Muthas, D.; Sabnis, Y.A.; Lundborg, M.; Karlén, A. Is It Possible to Increase Hit Rates in Structure-Based Virtual Screening by Pharmacophore Filtering? An Investigation of the Advantages and Pitfalls of Post-Filtering. J. Mol. Graph. Model. 2008, 26, 1237–1251.
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto Coefficient: Similarity Measures for Interaction Fingerprints. J. Cheminform. 2018, 10, 48.
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Implementing a Scoring Function Based on Interaction Fingerprint for Autogrow4: Protein Kinase CK1δ as a Case Study. Front. Mol. Biosci. 2022, 9, 629.
- Wang, H.; Wen, J.; Yang, Y.; Liu, H.; Wang, S.; Ding, X.; Zhou, C.; Zhang, X. Identification of Highly Effective Inhibitors against SARS-CoV-2 Main Protease: From Virtual Screening to in Vitro Study. Front. Pharmacol. 2022, 13, 4934.
- Tian, X.; Zhao, Q.; Chen, X.; Peng, Z.; Tan, X.; Wang, Q.; Chen, L.; Yang, Y. Discovery of Novel and Highly Potent Inhibitors of SARS-CoV-2 Papain-Like Protease Through Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, Molecular Dynamics Simulations, and Biological Evaluation. Front. Pharmacol. 2022, 13, 16.
- Yin, S.; Mei, S.; Li, Z.; Xu, Z.; Wu, Y.; Chen, X.; Liu, D.; Niu, M.-M.; Li, J. Non-Covalent Cyclic Peptides Simultaneously Targeting Mpro and NRP1 Are Highly Effective against Omicron BA.2.75. Front. Pharmacol. 2022, 13, 4723.
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A Blueprint for High Affinity SARS-CoV-2 Mpro Inhibitors from Activity-Based Compound Library Screening Guided by Analysis of Protein Dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095.
- Hu, X.; Chen, C.Z.; Xu, M.; Hu, Z.; Guo, H.; Itkin, Z.; Shinn, P.; Ivin, P.; Leek, M.; Liang, T.J.; et al. Discovery of Small Molecule Entry Inhibitors Targeting the Fusion Peptide of SARS-CoV-2 Spike Protein. ACS Med. Chem. Lett. 2021, 12, 1267–1274.
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs Repurposed for COVID-19 by Virtual Screening of 6,218 Drugs and Cell-Based Assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118.
- McGovern, S.L.; Shoichet, B.K. Information Decay in Molecular Docking Screens against Holo, Apo, and Modeled Conformations of Enzymes. J. Med. Chem. 2003, 46, 2895–2907.
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Combining Self- and Cross-Docking as Benchmark Tools: The Performance of DockBench in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 251–264.
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and Limitations of Ensemble Docking. J. Chem. Inf. Model. 2012, 52, 1262–1274.
- Knegtel, R.M.A.; Kuntz, I.D.; Oshiro, C.M. Molecular Docking to Ensembles of Protein Structures. J. Mol. Biol. 1997, 266, 424–440.
- Huang, S.Y.; Zou, X. Ensemble Docking of Multiple Protein Structures: Considering Protein Structural Variations in Molecular Docking. Proteins Struct. Funct. Genet. 2007, 66, 399–421.
- Wang, R.; Wang, S. How Does Consensus Scoring Work for Virtual Library Screening? An Idealized Computer Experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426.
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus Scoring: A Method for Obtaining Improved Hit Rates from Docking Databases of Three-Dimensional Structures into Proteins. J. Med. Chem. 1999, 42, 5100–5109.
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-Based Virtual Screening of Chemical Databases. 1. Evaluation of Different Docking/Scoring Combinations. J. Med. Chem. 2000, 43, 4759–4767.
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906.
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793.
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 3917–3926.
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216.
- Rubio-Martínez, J.; Jiménez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcón, D.; Vega, S.; Calvo, C.; Benítez, C.; Abian, O.; Velázquez-Campoy, A.; Thomson, T.M.; et al. Discovery of Diverse Natural Products as Inhibitors of SARS-CoV-2 MproProtease through Virtual Screening. J. Chem. Inf. Model. 2021, 61, 6094–6106.
- Clyde, A.; Galanie, S.; Kneller, D.W.; Ma, H.; Babuji, Y.; Blaiszik, B.; Brace, A.; Brettin, T.; Chard, K.; Chard, R.; et al. High-Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Noncovalent Inhibitor. J. Chem. Inf. Model. 2022, 62, 116–128.
- Glaab, E.; Manoharan, G.B.; Abankwa, D. Pharmacophore Model for SARS-CoV-2 3CLpro Small-Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors. J. Chem. Inf. Model. 2021, 61, 4082–4096.
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.H.; Cabeza De Vaca, I.; Liosi, M.E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533.
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703.
- Rastelli, G.; Degliesposti, G.; del Rio, A.; Sgobba, M. Binding Estimation after Refinement, a New Automated Procedure for the Refinement and Rescoring of Docked Ligands in Virtual Screening. Chem. Biol. Drug Des. 2009, 73, 283–286.
- Jespers, W.; Åqvist, J.; Gutiérrez-de-Terán, H. Free Energy Calculations for Protein–Ligand Binding Prediction. Methods Mol. Biol. 2021, 2266, 203–226.
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the Molecular Mechanics/Poisson Boltzmann Surface Area and Molecular Mechanics/Generalized Born Surface Area Methods. II. The Accuracy of Ranking Poses Generated from Docking. J. Comput. Chem. 2011, 32, 866–877.
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475.
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify Potent SARS-CoV-2 Main Protease Inhibitors via Accelerated Free Energy Perturbation-Based Virtual Screening of Existing Drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387.
- Ngo, S.T.; Tam, N.M.; Pham, M.Q.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 2302–2312.
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449.
- Ibrahim, I.M.; Elfiky, A.A.; Fathy, M.M.; Mahmoud, S.H.; ElHefnawi, M. Targeting SARS-CoV-2 Endoribonuclease: A Structure-Based Virtual Screening Supported by in Vitro Analysis. Sci. Rep. 2022, 12, 13337.
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739.
- Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of Drug Binding and Residence Time. Annu. Rev. Phys. Chem. 2019, 70, 143–171.
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Qualitative Estimation of Protein–Ligand Complex Stability through Thermal Titration Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 5715–5728.
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806.e5.
- Valiente, P.A.; Wen, H.; Nim, S.; Lee, J.; Kim, H.J.; Kim, J.; Perez-Riba, A.; Paudel, Y.P.; Hwang, I.; Kim, K.-D.; et al. Computational Design of Potent D-Peptide Inhibitors of SARS-CoV-2. J. Med. Chem. 2021, 64, 14955–14967.
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of Changes in Protein-Protein Binding Affinity on Mutations. Nucleic Acids Res. 2013, 41, W333–W339.
- Kandeel, M.; Yamamoto, M.; Tani, H.; Kobayashi, A.; Gohda, J.; Kawaguchi, Y.; Park, B.K.; Kwon, H.J.; Inoue, J.I.; Alkattan, A. Discovery of New Fusion Inhibitor Peptides against SARS-CoV-2 by Targeting the Spike S2 Subunit. Biomol. Ther. 2021, 29, 282–289.
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17, 665–680.
- Jeong, B.S.; Cha, J.S.; Hwang, I.; Kim, U.; Adolf-Bryfogle, J.; Coventry, B.; Cho, H.S.; Kim, K.D.; Oh, B.H. Computational Design of a Neutralizing Antibody with Picomolar Binding Affinity for All Concerning SARS-CoV-2 Variants. MAbs 2022, 14, 2021601.
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272.
- Dou, J.; Vorobieva, A.A.; Sheffler, W.; Doyle, L.A.; Park, H.; Bick, M.J.; Mao, B.; Foight, G.W.; Lee, M.Y.; Gagnon, L.A.; et al. De Novo Design of a Fluorescence-Activating β-Barrel. Nature 2018, 561, 485–491.
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De Novo Design of Picomolar SARS-CoV-2 Miniprotein Inhibitors. Science 2020, 370, 426–431.
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 2004, pl2.
- Kortemme, T.; Baker, D. A Simple Physical Model for Binding Energy Hot Spots in Protein-Protein Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121.
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 Receptor Traps Potently Neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055.
- de Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061.
- Ferrari, F.; Bissaro, M.; Fabbian, S.; de Almeida Roger, J.; Mammi, S.; Moro, S.; Bellanda, M.; Sturlese, M. HT-SuMD: Making Molecular Dynamics Simulations Suitable for Fragment-Based Screening. a Comparative Study with NMR. J. Enzym. Inhib. Med. Chem. 2020, 36, 1–14.
- Verdonk, M.L.; Giangreco, I.; Hall, R.J.; Korb, O.; Mortenson, P.N.; Murray, C.W. Docking Performance of Fragments and Druglike Compounds. J. Med. Chem. 2011, 54, 5422–5431.
- Bissaro, M.; Bolcato, G.; Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Inspecting the Mechanism of Fragment Hits Binding on SARS-CoV-2 M pro by Using Supervised Molecular Dynamics (SuMD) Simulations. ChemMedChem 2021, 16, 2075–2081.
- Sabbadin, D.; Moro, S. Supervised Molecular Dynamics (SuMD) as a Helpful Tool to Depict GPCR-Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 2014, 54, 372–376.
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705.
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the Mechanism of Action of SARS-CoV-2 Main Protease Inhibitor PF-07321332. J. Enzym. Inhib. Med. Chem. 2021, 36, 1646–1650.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Investigating RNA–Protein Recognition Mechanisms through Supervised Molecular Dynamics (SuMD) Simulations. NAR Genom. Bioinform. 2022, 4, lqac088.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
COVID-19
Revisions:
3 times
(View History)
Update Date:
08 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No