Since its outbreak in December 2019, the COVID-19 pandemic has caused the death of more than 6.5 million people around the world. The high transmissibility of its causative agent, the SARS-CoV-2 virus, coupled with its potentially lethal outcome, provoked a profound global economic and social crisis. The urgency of finding suitable pharmacological tools to tame the pandemic shed light on the ever-increasing importance of computer simulations in rationalizing and speeding up the design of new drugs, further stressing the need for developing quick and reliable methods to identify novel active molecules and characterize their mechanism of action.
- COVID-19
- SARS-CoV-2
- rational drug design
- CADD
- SBDD
- homology modeling
- docking
- pharmacophore
- protein–ligand interaction fingerprints
- molecular dynamics
1. Rational Design of COVID-19 Drugs
2. Computer Simulations for Rational Drug Design
2.1. CADD Strategies against COVID-19
2.2. The Swiss Knife of SBDD: Molecular Docking
2.3. Complementary Strategies to Address Docking Limitations
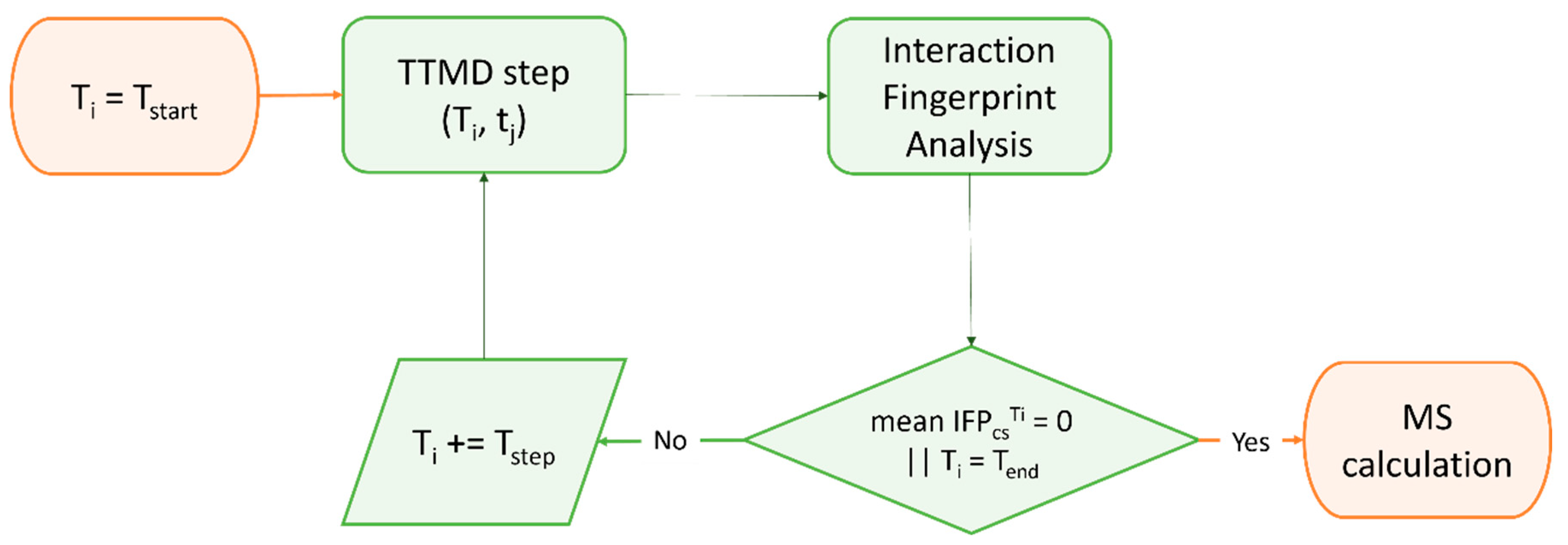
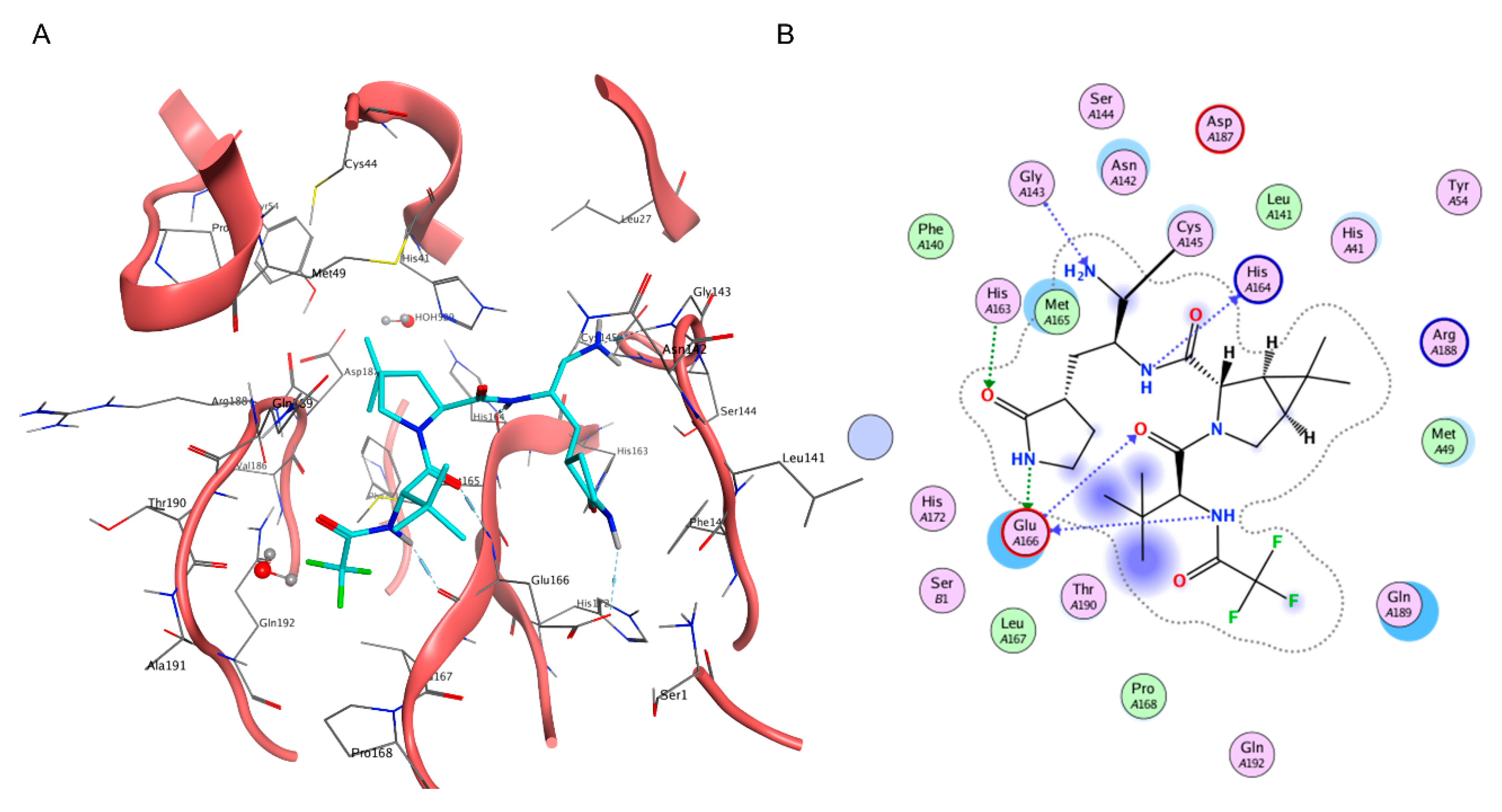
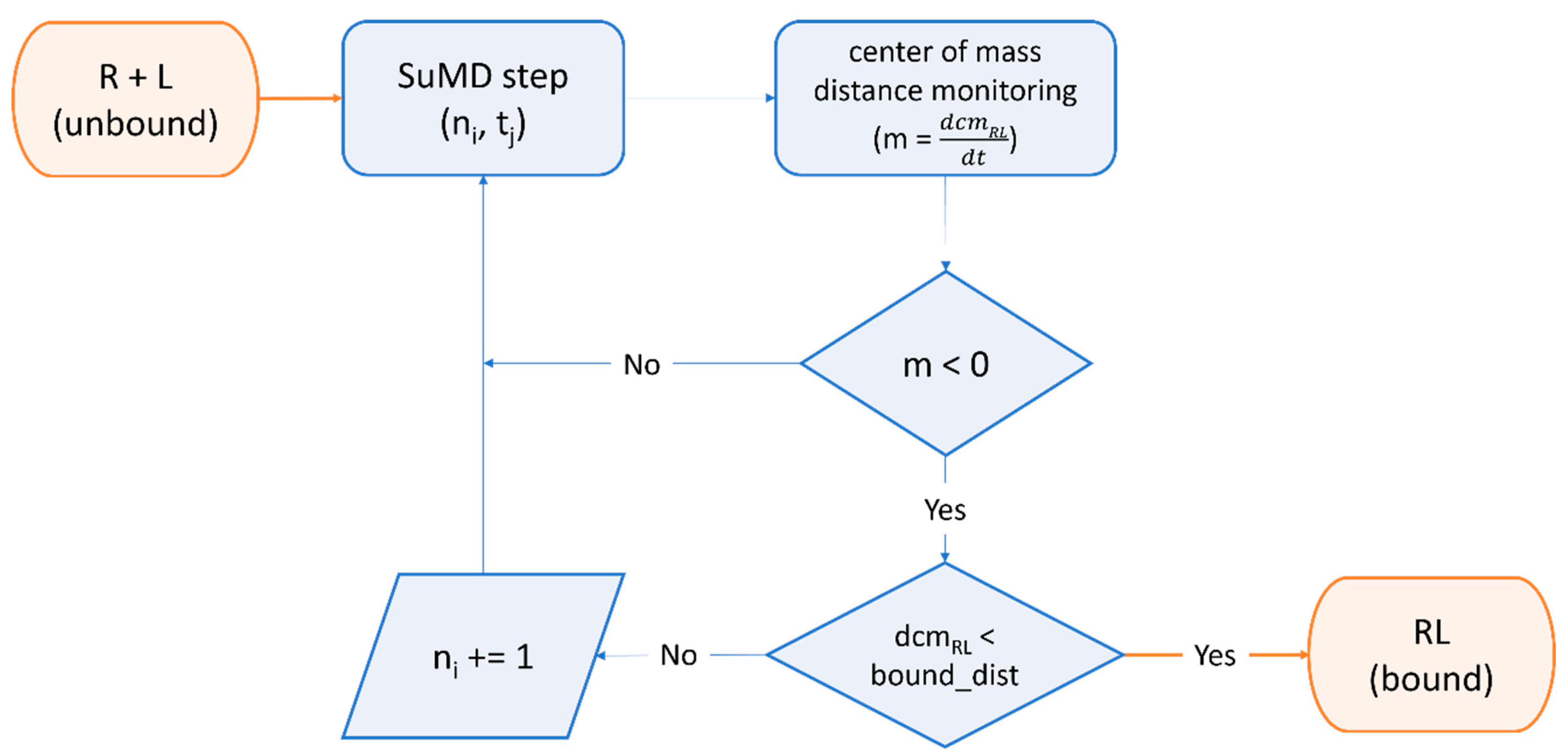
2.4. Beyond Protein-Ligand Docking: Alternative Strategies for Rational Drug Development
This entry is adapted from the peer-reviewed paper 10.3390/ijms24054401
References
- de Clercq, E. Strategies in the Design of Antiviral Drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25.
- Kilianski, A.; Baker, S.C. Cell-Based Antiviral Screening against Coronaviruses: Developing Virus-Specific and Broad-Spectrum Inhibitors. Antiviral Res. 2014, 101, 105–112.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242.
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797.
- Njoroge, F.G.; Chen, K.X.; Shih, N.Y.; Piwinski, J.J. Challenges in Modern Drug Discovery: A Case Study of Boceprevir, an HCV Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. Acc. Chem. Res. 2008, 41, 50–59.
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From Non-A, Non-B Hepatitis to Hepatitis C Virus Cure. J. Hepatol. 2015, 62, S87–S99.
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 Protease: A Major Success of Structure-Assisted Drug Design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284.
- Palella, F.J.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998, 338, 853–860.
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.E.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small Molecules Targeting Severe Acute Respiratory Syndrome Human Coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017.
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293.
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What Coronavirus 3C-like Protease Tells Us: From Structure, Substrate Selectivity, to Inhibitor Design. Med. Res. Rev. 2021, 41, 1965–1998.
- Poordad, F.; McCone, J.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for Untreated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1195–1206.
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1207–1217.
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like Protease Inhibitor in Treating Various Forms of acquired Feline Infectious Peritonitis. J. Feline Med. Surg. 2018, 20, 378.
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692.
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282.
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628.
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747.
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical Characterization of an Intravenous Coronavirus 3CL Protease Inhibitor for the Potential Treatment of COVID19. Nat. Commun. 2021, 12, 6055.
- De Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A Comparative Analysis of SARS-CoV-2 Antivirals Characterizes 3CLpro Inhibitor PF-00835231 as a Potential New Treatment for COVID-19. J. Virol. 2021, 95, e01819-20.
- Vandyck, K.; Deval, J. Considerations for the Discovery and Development of 3-Chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 2021, 49, 36.
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 M pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26.
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341.
- How Pfizer Scientists Transformed an Old Drug Lead into a COVID-19 Antiviral. Available online: https://cen.acs.org/pharmaceuticals/drug-discovery/How-Pfizer-scientists-transformed-an-old-drug-lead-into-a-COVID-19-antiviral/100/i3 (accessed on 14 December 2022).
- Pfizer Unveils Its Oral SARS-CoV-2 Inhibitor. Available online: https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13 (accessed on 14 December 2022).
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591.
- Mahase, E. COVID-19: Pfizer’s Paxlovid Is 89% Effective in Patients at Risk of Serious Illness, Company Reports. BMJ 2021, 375, n2713.
- Hung, Y.P.; Lee, J.C.; Chiu, C.W.; Lee, C.C.; Tsai, P.J.; Hsu, I.L.; Ko, W.C. Oral Nirmatrelvir/Ritonavir Therapy for COVID-19: The Dawn in the Dark? Antibiotics 2022, 11, 220.
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main Protease Mutants of SARS-CoV-2 Variants Remain Susceptible to Nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629.
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D.; et al. SARS-CoV-2 3CLpro Mutations Selected in a VSV-Based System Confer Resistance to Nirmatrelvir, Ensitrelvir, and GC376. Sci. Transl. Med. 2022, 15, eabq7360.
- McCarthy, M.W. Ensitrelvir as a Potential Treatment for COVID-19. Expert Opin. Pharmacother. 2022, 23, 1995–1998.
- Shimizu, R.; Sonoyama, T.; Fukuhara, T.; Kuwata, A.; Matsuo, Y.; Kubota, R. Safety, Tolerability, and Pharmacokinetics of the Novel Antiviral Agent Ensitrelvir Fumaric Acid, a SARS-CoV-2 3CL Protease Inhibitor, in Healthy Adults. Antimicrob. Agents Chemother. 2022, 66, e00632-22.
- Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; et al. A Randomized Phase 2/3 Study of Ensitrelvir, a Novel Oral SARS-CoV-2 3C-Like Protease Inhibitor, in Japanese Patients with Mild-to-Moderate COVID-19 or Asymptomatic SARS-CoV-2 Infection: Results of the Phase 2a Part. Antimicrob. Agents Chemother. 2022, 66, e00697-22.
- Cragg, G.M.; Newman, D.J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695.
- Gurib-Fakim, A. Medicinal Plants: Traditions of Yesterday and Drugs of Tomorrow. Mol. Asp. Med. 2006, 27, 1–93.
- Harvey, A.L. Natural Products in Drug Discovery. Drug Discov. Today 2008, 13, 894–901.
- Ban, T.A. The Role of Serendipity in Drug Discovery. Dialogues Clin. Neurosci. 2006, 8, 335–344.
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651.
- Jaskolski, M.; Dauter, Z.; Wlodawer, A. A Brief History of Macromolecular Crystallography, Illustrated by a Family Tree and Its Nobel Fruits. FEBS J. 2014, 281, 3985–4009.
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395.
- Leelananda, S.P.; Lindert, S. Computational Methods in Drug Discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718.
- Kapetanovic, I.M. Computer-Aided Drug Discovery and Development (CADDD): In Silico-Chemico-Biological Approach. Chem. Biol. Interact. 2008, 171, 165–176.
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharm. Res. 2015, 38, 1686–1701.
- Yu, W.; Mackerell, A.D. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106.
- Bai, X.C.; McMullan, G.; Scheres, S.H.W. How Cryo-EM Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589.
- Smyth, M.S.; Martin, J.H.J. X Ray Crystallography. Mol. Pathol. 2000, 53, 8.
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, e1000168.
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) Is the Limit. Int. J. Mol. Sci. 2019, 20, 4186.
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697.
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325.
- Cavasotto, C.N.; Phatak, S.S. Homology Modeling in Drug Discovery: Current Trends and Applications. Drug Discov. Today 2009, 14, 676–683.
- Bonneau, R.; Strauss, C.E.M.; Rohl, C.A.; Chivian, D.; Bradley, P.; Malmströ, L.; Robertson, T.; Baker, D. De Novo Prediction of Three-Dimensional Structures for Major Protein Families. J. Mol. Biol. 2002, 322, 65–78.
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148.
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-Accuracy Protein Structure Prediction in CASP14. Proteins 2021, 89, 1687–1699.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1711–1721.
- Kwon, S.; Won, J.; Kryshtafovych, A.; Seok, C. Assessment of Protein Model Structure Accuracy Estimation in CASP14: Old and New Challenges. Proteins Struct. Funct. Bioinform. 2021, 89, 1940–1948.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. From the Wuhan-Hu-1 Strain to the XD and XE Variants: Is Targeting the SARS-CoV-2 Spike Protein Still a Pharmaceutically Relevant Option against COVID-19? J. Enzyme Inhib. Med. Chem. 2022, 37, 1704–1714.
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K.; et al. Evolution of the SARS-CoV-2 Proteome in Three Dimensions (3D) during the First 6 Months of the COVID-19 Pandemic. Proteins: Struct. Funct. Bioinform. 2022, 90, 1054–1080.
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.L.; Li, J. A Guideline for Homology Modeling of the Proteins from Newly Discovered Betacoronavirus, 2019 Novel Coronavirus (2019-NCoV). J. Med. Virol. 2020, 92, 1542–1548.
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788.
- Bassani, D.; Ragazzi, E.; Lapolla, A.; Sartore, G.; Moro, S. Omicron Variant of SARS-CoV-2 Virus: In Silico Evaluation of the Possible Impact on People Affected by Diabetes Mellitus. Front. Endocrinol. 2022, 13, 284.
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface Can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol. Biol. 2021, 433, 167051.
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601.e6.
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654.
- Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation Signatures and in Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Bat Coronaviruses Related to SARS-CoV-2: What about Their 3CL Proteases (MPro)? J. Enzym. Inhib. Med. Chem. 2022, 37, 1077–1082.
- Martin, R.W.; Butts, C.T.; Cross, T.J.; Takahashi, G.R.; Diessner, E.M.; Crosby, M.G.; Farahmand, V.; Zhuang, S. Sequence Characterization and Molecular Modeling of Clinically Relevant Variants of the SARS-CoV-2 Main Protease. Biochemistry 2020, 59, 3741–3756.
- Huang, X.; Zhang, C.; Pearce, R.; Omenn, G.S.; Zhang, Y. Identifying the Zoonotic Origin of SARS-CoV-2 by Modeling the Binding Affinity between the Spike Receptor-Binding Domain and Host ACE2. J. Proteome Res. 2020, 19, 4844–4856.
- Rodrigues, J.P.G.L.M.; Barrera-Vilarmau, S.; Teixeira, J.M.C.; Sorokina, M.; Seckel, E.; Kastritis, P.L.; Levitt, M. Insights on Cross-Species Transmission of SARS-CoV-2 from Structural Modeling. PLoS Comput. Biol. 2020, 16, e1008449.
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In Silico Comparison of SARS-CoV-2 Spike Protein-ACE2 Binding Affinities across Species and Implications for Virus Origin. Sci. Rep. 2021, 11, 13063.
- Sharma, P.; Kumar, M.; Tripathi, M.K.; Gupta, D.; Vishwakarma, P.; Das, U.; Kaur, P. Genomic and Structural Mechanistic Insight to Reveal the Differential Infectivity of Omicron and Other Variants of Concern. Comput. Biol. Med. 2022, 150, 106129.
- Jacob, J.J.; Vasudevan, K.; Pragasam, A.K.; Gunasekaran, K.; Veeraraghavan, B.; Mutreja, A. Evolutionary Tracking of SARS-CoV-2 Genetic Variants Highlights an Intricate Balance of Stabilizing and Destabilizing Mutations. mBio 2021, 12, e01188-21.
- Luo, R.; Delaunay-Moisan, A.; Timmis, K.; Danchin, A. SARS-CoV-2 Biology and Variants: Anticipation of Viral Evolution and What Needs to Be Done. Environ. Microbiol. 2021, 23, 2339–2363.
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and COVID-19 Therapeutics. ChemMedChem 2020, 15, 907–932.
- Ghosh, A.K.; Mishevich, J.L.; Mesecar, A.; Mitsuya, H. Recent Drug Development and Medicinal Chemistry Approaches for the Treatment of SARS-CoV-2 Infection and COVID-19. ChemMedChem 2022, 17, e202200440.
- Tiwari, V.; Beer, J.C.; Sankaranarayanan, N.V.; Swanson-Mungerson, M.; Desai, U.R. Discovering Small-Molecule Therapeutics against SARS-CoV-2. Drug Discov. Today 2020, 25, 1535–1544.
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral Drug Discovery: Preparing for the next Pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655.
- Consortium, T.C.M.; Achdout, H.; Aimon, A.; Bar-David, E.; Barr, H.; Ben-Shmuel, A.; Bennett, J.; Bilenko, V.A.; Bilenko, V.A.; Boby, M.L.; et al. Open Science Discovery of Oral Non-Covalent SARS-CoV-2 Main Protease Inhibitor Therapeutics. bioRxiv 2022.
- Kuntz, I.D. Structure-Based Strategies for Drug Design and Discovery. Science 1992, 257, 1078–1082.
- Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732–18758.
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389.
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of Protein Pockets and Cavities: Measurement of Binding Site Geometry and Implications for Ligand Design. Protein Sci. 1998, 7, 1884–1897.
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discov. 2004, 3, 935–949.
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. J. Mol. Biol. 1982, 161, 269–288.
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Current Computer Aided-Drug Design 2012, 7, 146–157.
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923.
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of Docking: An Overview of Search Algorithms and a Guide to Scoring Functions. Proteins Struct. Funct. Genet. 2002, 47, 409–443.
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931.
- Bassani, D.; Pavan, M.; Bolcato, G.; Sturlese, M.; Moro, S. Re-Exploring the Ability of Common Docking Programs to Correctly Reproduce the Binding Modes of Non-Covalent Inhibitors of SARS-CoV-2 Protease Mpro. Pharmaceuticals 2022, 15, 180.
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028.
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to COVID-19. J. Chem. Inf. Model. 2020, 60, 5832–5852.
- Gorgulla, C.; Padmanabha Das, K.M.; Leigh, K.E.; Cespugli, M.; Fischer, P.D.; Wang, Z.F.; Tesseyre, G.; Pandita, S.; Shnapir, A.; Calderaio, A.; et al. A Multi-Pronged Approach Targeting SARS-CoV-2 Proteins Using Ultra-Large Virtual Screening. iScience 2021, 24, 102021.
- Manelfi, C.; Gossen, J.; Gervasoni, S.; Talarico, C.; Albani, S.; Philipp, B.J.; Musiani, F.; Vistoli, G.; Rossetti, G.; Beccari, A.R.; et al. Combining Different Docking Engines and Consensus Strategies to Design and Validate Optimized Virtual Screening Protocols for the SARS-CoV-2 3CL Protease. Molecules 2021, 26, 797.
- Corona, A.; Wycisk, K.; Talarico, C.; Manelfi, C.; Milia, J.; Cannalire, R.; Esposito, F.; Gribbon, P.; Zaliani, A.; Iaconis, D.; et al. Natural Compounds Inhibit SARS-CoV-2 Nsp13 Unwinding and ATPase Enzyme Activities. ACS Pharmacol. Transl. Sci. 2022, 5, 226–239.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins 2003, 52, 609–623.
- Kolarič, A.; Jukič, M.; Bren, U. Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1. Pharmaceuticals 2022, 15, 165.
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785.
- Vatansever, E.C.; Yang, K.S.; Drelich, A.K.; Kratch, K.C.; Cho, C.C.; Kempaiah, K.R.; Hsu, J.C.; Mellott, D.M.; Xu, S.; Tseng, C.T.K.; et al. Bepridil Is Potent against SARS-CoV-2 in Vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2012201118.
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and Scoring with ICM: The Benchmarking Results and Strategies for Improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686.
- Lam, P.C.H.; Abagyan, R.; Totrov, M. Ligand-Biased Ensemble Receptor Docking (LigBEnD): A Hybrid Ligand/Receptor Structure-Based Approach. J. Comput. Aided Mol. Des. 2018, 32, 187–198.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455.
- Kao, H.T.; Orry, A.; Palfreyman, M.G.; Porton, B. Synergistic Interactions of Repurposed Drugs That Inhibit Nsp1, a Major Virulence Factor for COVID-19. Sci. Rep. 2022, 12, 10174.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749.
- Zhang, Y.; Gao, H.; Hu, X.; Wang, Q.; Zhong, F.; Zhou, X.; Lin, C.; Yang, Y.; Wei, J.; Du, W.; et al. Structure-Based Discovery and Structural Basis of a Novel Broad-Spectrum Natural Product against the Main Protease of Coronavirus. J. Virol. 2022, 96, 1253–1274.
- Huff, S.; Kummetha, I.R.; Tiwari, S.K.; Huante, M.B.; Clark, A.E.; Wang, S.; Bray, W.; Smith, D.; Carlin, A.F.; Endsley, M.; et al. Discovery and Mechanism of SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2866–2879.
- Liu, K.; Zou, R.; Cui, W.; Li, M.; Wang, X.; Dong, J.; Li, H.; Li, H.; Wang, P.; Shao, X.; et al. Clinical HDAC Inhibitors Are Effective Drugs to Prevent the Entry of SARS-CoV2. ACS Pharmacol. Transl. Sci. 2020, 3, 1361–1370.
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation Studies of the Site-Directed Docking Program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171.
- Wang, G.; Yang, M.L.; Duan, Z.L.; Liu, F.L.; Jin, L.; Long, C.B.; Zhang, M.; Tang, X.P.; Xu, L.; Li, Y.C.; et al. Dalbavancin Binds ACE2 to Block Its Interaction with SARS-CoV-2 Spike Protein and Is Effective in Inhibiting SARS-CoV-2 Infection in Animal Models. Cell Res. 2020, 31, 17–24.
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; de Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc 2022, 144, 2905–2920.
- Cross, S.S.J. Improved FlexX Docking Using FlexS-Determined Base Fragment Placement. J. Chem. Inf. Model. 2005, 45, 993–1001.
- Welker, A.; Kersten, C.; Müller, C.; Madhugiri, R.; Zimmer, C.; Müller, P.; Zimmermann, R.; Hammerschmidt, S.; Maus, H.; Ziebuhr, J.; et al. Structure-Activity Relationships of Benzamides and Isoindolines Designed as SARS-CoV Protease Inhibitors Effective against SARS-CoV-2. ChemMedChem 2021, 16, 340–354.
- Otava, T.; Šála, M.; Li, F.; Fanfrlík, J.; Devkota, K.; Perveen, S.; Chau, I.; Pakarian, P.; Hobza, P.; Vedadi, M.; et al. The Structure-Based Design of SARS-CoV-2 Nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors. ACS Infect Dis. 2021, 7, 2214–2220.
- Wang, Y.T.; Long, X.Y.; Ding, X.; Fan, S.R.; Cai, J.Y.; Yang, B.J.; Zhang, X.F.; Luo, R.H.; Yang, L.; Ruan, T.; et al. Novel Nucleocapsid Protein-Targeting Phenanthridine Inhibitors of SARS-CoV-2. Eur. J. Med. Chem. 2022, 227, 113966.
- Chen, Y.C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95.
- Llanos, M.A.; Gantner, M.E.; Rodriguez, S.; Alberca, L.N.; Bellera, C.L.; Talevi, A.; Gavernet, L. Strengths and Weaknesses of Docking Simulations in the SARS-CoV-2 Era: The Main Protease (Mpro) Case Study. J. Chem. Inf. Model. 2021, 61, 3758–3770.
- Chaput, L.; Mouawad, L. Efficient Conformational Sampling and Weak Scoring in Docking Programs? Strategy of the Wisdom of Crowds. J. Cheminform. 2017, 9, 37.
- Neves, B.J.; Mottin, M.; Moreira-Filho, J.T.; de Paula Sousa, B.K.; Mendonca, S.S.; Andrade, C.H. Best Practices for Docking-Based Virtual Screening. Mol. Docking Comput. Aided Drug Des. 2021, 2021, 75–98.
- Cerón-Carrasco, J.P. When Virtual Screening Yields Inactive Drugs: Dealing with False Theoretical Friends. ChemMedChem 2022, 17, e202200278.
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881.
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining Docking and Molecular Dynamic Simulations in Drug Design. Med. Res. Rev. 2006, 26, 531–568.
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129.
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652.
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 1–9.
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421.
- Tan, L.; Batista, J.; Bajorath, J. Computational Methodologies for Compound Database Searching That Utilize Experimental Protein-Ligand Interaction Information. Chem. Biol. Drug Des. 2010, 76, 191–200.
- Peach, M.L.; Nicklaus, M.C. Combining Docking with Pharmacophore Filtering for Improved Virtual Screening. J. Cheminform. 2009, 1, 6.
- Muthas, D.; Sabnis, Y.A.; Lundborg, M.; Karlén, A. Is It Possible to Increase Hit Rates in Structure-Based Virtual Screening by Pharmacophore Filtering? An Investigation of the Advantages and Pitfalls of Post-Filtering. J. Mol. Graph. Model. 2008, 26, 1237–1251.
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto Coefficient: Similarity Measures for Interaction Fingerprints. J. Cheminform. 2018, 10, 48.
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Implementing a Scoring Function Based on Interaction Fingerprint for Autogrow4: Protein Kinase CK1δ as a Case Study. Front. Mol. Biosci. 2022, 9, 629.
- Wang, H.; Wen, J.; Yang, Y.; Liu, H.; Wang, S.; Ding, X.; Zhou, C.; Zhang, X. Identification of Highly Effective Inhibitors against SARS-CoV-2 Main Protease: From Virtual Screening to in Vitro Study. Front. Pharmacol. 2022, 13, 4934.
- Tian, X.; Zhao, Q.; Chen, X.; Peng, Z.; Tan, X.; Wang, Q.; Chen, L.; Yang, Y. Discovery of Novel and Highly Potent Inhibitors of SARS-CoV-2 Papain-Like Protease Through Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, Molecular Dynamics Simulations, and Biological Evaluation. Front. Pharmacol. 2022, 13, 16.
- Yin, S.; Mei, S.; Li, Z.; Xu, Z.; Wu, Y.; Chen, X.; Liu, D.; Niu, M.-M.; Li, J. Non-Covalent Cyclic Peptides Simultaneously Targeting Mpro and NRP1 Are Highly Effective against Omicron BA.2.75. Front. Pharmacol. 2022, 13, 4723.
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A Blueprint for High Affinity SARS-CoV-2 Mpro Inhibitors from Activity-Based Compound Library Screening Guided by Analysis of Protein Dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095.
- Hu, X.; Chen, C.Z.; Xu, M.; Hu, Z.; Guo, H.; Itkin, Z.; Shinn, P.; Ivin, P.; Leek, M.; Liang, T.J.; et al. Discovery of Small Molecule Entry Inhibitors Targeting the Fusion Peptide of SARS-CoV-2 Spike Protein. ACS Med. Chem. Lett. 2021, 12, 1267–1274.
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs Repurposed for COVID-19 by Virtual Screening of 6,218 Drugs and Cell-Based Assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118.
- McGovern, S.L.; Shoichet, B.K. Information Decay in Molecular Docking Screens against Holo, Apo, and Modeled Conformations of Enzymes. J. Med. Chem. 2003, 46, 2895–2907.
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Combining Self- and Cross-Docking as Benchmark Tools: The Performance of DockBench in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 251–264.
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and Limitations of Ensemble Docking. J. Chem. Inf. Model. 2012, 52, 1262–1274.
- Knegtel, R.M.A.; Kuntz, I.D.; Oshiro, C.M. Molecular Docking to Ensembles of Protein Structures. J. Mol. Biol. 1997, 266, 424–440.
- Huang, S.Y.; Zou, X. Ensemble Docking of Multiple Protein Structures: Considering Protein Structural Variations in Molecular Docking. Proteins Struct. Funct. Genet. 2007, 66, 399–421.
- Wang, R.; Wang, S. How Does Consensus Scoring Work for Virtual Library Screening? An Idealized Computer Experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426.
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus Scoring: A Method for Obtaining Improved Hit Rates from Docking Databases of Three-Dimensional Structures into Proteins. J. Med. Chem. 1999, 42, 5100–5109.
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-Based Virtual Screening of Chemical Databases. 1. Evaluation of Different Docking/Scoring Combinations. J. Med. Chem. 2000, 43, 4759–4767.
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906.
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793.
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 3917–3926.
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216.
- Rubio-Martínez, J.; Jiménez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcón, D.; Vega, S.; Calvo, C.; Benítez, C.; Abian, O.; Velázquez-Campoy, A.; Thomson, T.M.; et al. Discovery of Diverse Natural Products as Inhibitors of SARS-CoV-2 MproProtease through Virtual Screening. J. Chem. Inf. Model. 2021, 61, 6094–6106.
- Clyde, A.; Galanie, S.; Kneller, D.W.; Ma, H.; Babuji, Y.; Blaiszik, B.; Brace, A.; Brettin, T.; Chard, K.; Chard, R.; et al. High-Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Noncovalent Inhibitor. J. Chem. Inf. Model. 2022, 62, 116–128.
- Glaab, E.; Manoharan, G.B.; Abankwa, D. Pharmacophore Model for SARS-CoV-2 3CLpro Small-Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors. J. Chem. Inf. Model. 2021, 61, 4082–4096.
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.H.; Cabeza De Vaca, I.; Liosi, M.E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533.
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703.
- Rastelli, G.; Degliesposti, G.; del Rio, A.; Sgobba, M. Binding Estimation after Refinement, a New Automated Procedure for the Refinement and Rescoring of Docked Ligands in Virtual Screening. Chem. Biol. Drug Des. 2009, 73, 283–286.
- Jespers, W.; Åqvist, J.; Gutiérrez-de-Terán, H. Free Energy Calculations for Protein–Ligand Binding Prediction. Methods Mol. Biol. 2021, 2266, 203–226.
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the Molecular Mechanics/Poisson Boltzmann Surface Area and Molecular Mechanics/Generalized Born Surface Area Methods. II. The Accuracy of Ranking Poses Generated from Docking. J. Comput. Chem. 2011, 32, 866–877.
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475.
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify Potent SARS-CoV-2 Main Protease Inhibitors via Accelerated Free Energy Perturbation-Based Virtual Screening of Existing Drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387.
- Ngo, S.T.; Tam, N.M.; Pham, M.Q.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 2302–2312.
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449.
- Ibrahim, I.M.; Elfiky, A.A.; Fathy, M.M.; Mahmoud, S.H.; ElHefnawi, M. Targeting SARS-CoV-2 Endoribonuclease: A Structure-Based Virtual Screening Supported by in Vitro Analysis. Sci. Rep. 2022, 12, 13337.
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739.
- Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of Drug Binding and Residence Time. Annu. Rev. Phys. Chem. 2019, 70, 143–171.
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Qualitative Estimation of Protein–Ligand Complex Stability through Thermal Titration Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 5715–5728.
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806.e5.
- Valiente, P.A.; Wen, H.; Nim, S.; Lee, J.; Kim, H.J.; Kim, J.; Perez-Riba, A.; Paudel, Y.P.; Hwang, I.; Kim, K.-D.; et al. Computational Design of Potent D-Peptide Inhibitors of SARS-CoV-2. J. Med. Chem. 2021, 64, 14955–14967.
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of Changes in Protein-Protein Binding Affinity on Mutations. Nucleic Acids Res. 2013, 41, W333–W339.
- Kandeel, M.; Yamamoto, M.; Tani, H.; Kobayashi, A.; Gohda, J.; Kawaguchi, Y.; Park, B.K.; Kwon, H.J.; Inoue, J.I.; Alkattan, A. Discovery of New Fusion Inhibitor Peptides against SARS-CoV-2 by Targeting the Spike S2 Subunit. Biomol. Ther. 2021, 29, 282–289.
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17, 665–680.
- Jeong, B.S.; Cha, J.S.; Hwang, I.; Kim, U.; Adolf-Bryfogle, J.; Coventry, B.; Cho, H.S.; Kim, K.D.; Oh, B.H. Computational Design of a Neutralizing Antibody with Picomolar Binding Affinity for All Concerning SARS-CoV-2 Variants. MAbs 2022, 14, 2021601.
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272.
- Dou, J.; Vorobieva, A.A.; Sheffler, W.; Doyle, L.A.; Park, H.; Bick, M.J.; Mao, B.; Foight, G.W.; Lee, M.Y.; Gagnon, L.A.; et al. De Novo Design of a Fluorescence-Activating β-Barrel. Nature 2018, 561, 485–491.
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De Novo Design of Picomolar SARS-CoV-2 Miniprotein Inhibitors. Science 2020, 370, 426–431.
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 2004, pl2.
- Kortemme, T.; Baker, D. A Simple Physical Model for Binding Energy Hot Spots in Protein-Protein Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121.
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 Receptor Traps Potently Neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055.
- de Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061.
- Ferrari, F.; Bissaro, M.; Fabbian, S.; de Almeida Roger, J.; Mammi, S.; Moro, S.; Bellanda, M.; Sturlese, M. HT-SuMD: Making Molecular Dynamics Simulations Suitable for Fragment-Based Screening. a Comparative Study with NMR. J. Enzym. Inhib. Med. Chem. 2020, 36, 1–14.
- Verdonk, M.L.; Giangreco, I.; Hall, R.J.; Korb, O.; Mortenson, P.N.; Murray, C.W. Docking Performance of Fragments and Druglike Compounds. J. Med. Chem. 2011, 54, 5422–5431.
- Bissaro, M.; Bolcato, G.; Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Inspecting the Mechanism of Fragment Hits Binding on SARS-CoV-2 M pro by Using Supervised Molecular Dynamics (SuMD) Simulations. ChemMedChem 2021, 16, 2075–2081.
- Sabbadin, D.; Moro, S. Supervised Molecular Dynamics (SuMD) as a Helpful Tool to Depict GPCR-Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 2014, 54, 372–376.
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705.
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the Mechanism of Action of SARS-CoV-2 Main Protease Inhibitor PF-07321332. J. Enzym. Inhib. Med. Chem. 2021, 36, 1646–1650.
- Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Investigating RNA–Protein Recognition Mechanisms through Supervised Molecular Dynamics (SuMD) Simulations. NAR Genom. Bioinform. 2022, 4, lqac088.