 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martha Patricia Sierra-Vargas | -- | 2898 | 2023-02-01 05:36:52 | | | |
| 2 | Jessie Wu | Meta information modification | 2898 | 2023-02-02 01:56:43 | | | | |
| 3 | Jessie Wu | Meta information modification | 2898 | 2023-02-02 02:01:27 | | |
Video Upload Options
Redox regulation participates in the control of various aspects of metabolism. Reactive oxygen and nitrogen species participate in many reactions under physiological conditions. When these species overcome the antioxidant defense system, a distressed status emerges, increasing biomolecular damage and leading to functional alterations. Air pollution is one of the exogenous sources of reactive oxygen and nitrogen species. Ambient airborne particulate matter (PM) is important because of its complex composition, which includes transition metals and organic compounds. Once in contact with the lungs’ epithelium, PM components initiate the synthesis of inflammatory mediators, macrophage activation, modulation of gene expression, and the activation of transcription factors, which are all related to the physiopathology of chronic respiratory diseases, including cancer.
1. Introduction
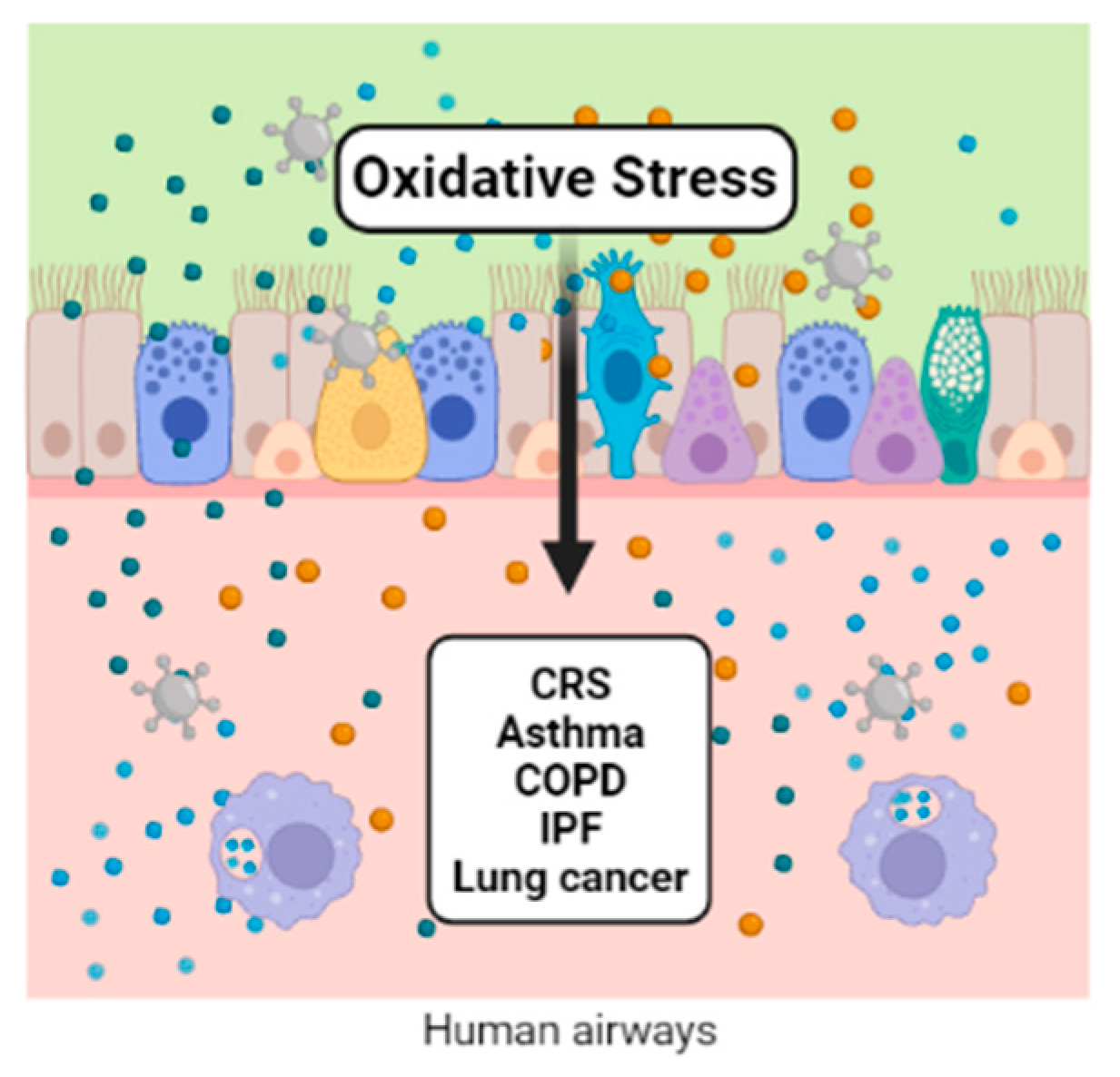
2. Chronic Rhinosinusitis (CRS) and Nasal Polyps (NP)
3. Asthma
References
- Pruchniak, M.P.; Araźna, M.; Demkow, U. Biochemistry of Oxidative Stress BT—Advances in Clinical Science; Pokorski, M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 9–19. ISBN 978-3-319-21497-9.
- Kruk, J.; Aboul-Enein, H.Y.; Kładna, A.; Bowser, J.E. Oxidative Stress in Biological Systems and Its Relation with Pathophysiological Functions: The Effect of Physical Activity on Cellular Redox Homeostasis. Free Radic. Res. 2019, 53, 497–521.
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol. Cell. Biochem. 2020, 467, 1–12.
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763.
- Mudway, I.S.; Kelly, F.J.; Holgate, S.T. Oxidative Stress in Air Pollution Research. Free Radic. Biol. Med. 2020, 151, 2–6.
- Sedaghat, A.R.; Kuan, E.C.; Scadding, G.K. Epidemiology of Chronic Rhinosinusitis: Prevalence and Risk Factors. J. Allergy Clin. Immunol. Pract. 2022, 10, 1395–1403.
- Patel, T.R.; Tajudeen, B.A.; Brown, H.; Gattuso, P.; LoSavio, P.; Papagiannopoulos, P.; Batra, P.S.; Mahdavinia, M. Association of Air Pollutant Exposure and Sinonasal Histopathology Findings in Chronic Rhinosinusitis. Am. J. Rhinol. Allergy 2021, 35, 761–767.
- Velasquez, N.; Gardiner, L.; Cheng, T.Z.; Moore, J.A.; Boudreau, R.M.; Presto, A.A.; Lee, S.E. Relationship between Socioeconomic Status, Exposure to Airborne Pollutants, and Chronic Rhinosinusitis Disease Severity. Int. Forum Allergy Rhinol. 2022, 12, 172–180.
- Velasquez, N.; Moore, J.A.; Boudreau, R.M.; Mady, L.J.; Lee, S.E. Association of Air Pollutants, Airborne Occupational Exposures, and Chronic Rhinosinusitis Disease Severity. Int. Forum Allergy Rhinol. 2020, 10, 175–182.
- Zheng, K.; Hao, J.; Xiao, L.; Wang, M.; Zhao, Y.; Fan, D.; Li, Y.; Wang, X.; Zhang, L. Expression of Nicotinamide Adenine Dinucleotide Phosphate Oxidase in Chronic Rhinosinusitis with Nasal Polyps. Int. Forum Allergy Rhinol. 2020, 10, 646–655.
- Mihalj, H.; Butković, J.; Tokić, S.; Štefanić, M.; Kizivat, T.; Bujak, M.; Baus Lončar, M.; Mihalj, M. Expression of Oxidative Stress and Inflammation-Related Genes in Nasal Mucosa and Nasal Polyps from Patients with Chronic Rhinosinusitis. Int. J. Mol. Sci. 2022, 23, 5521.
- Zorlu, M.E.; Uygur, K.K.; Yılmaz, N.S.; Demirel, Ö.Ö.; Aydil, U.; Kızıl, Y.; Uslu, S. Evaluation of Advanced Oxidation Protein Products (AOPP) and Superoxide Dismutase (SOD) Tissue Levels in Patients with Nasal Polyps. Indian J. Otolaryngol. Head Neck Surg. 2022.
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103.
- Lin, H.; Ba, G.; Tang, R.; Li, M.; Li, Z.; Li, D.; Ye, H.; Zhang, W. Increased Expression of TXNIP Facilitates Oxidative Stress in Nasal Epithelial Cells of Patients With Chronic Rhinosinusitis With Nasal Polyps. Am. J. Rhinol. Allergy 2021, 35, 607–614.
- Ramanathan, M.J.; Tharakan, A.; Sidhaye, V.K.; Lane, A.P.; Biswal, S.; London, N.R.J. Disruption of Sinonasal Epithelial Nrf2 Enhances Susceptibility to Rhinosinusitis in a Mouse Model. Laryngoscope 2021, 131, 713–719.
- London, N.R.J.; Tharakan, A.; Mendiola, M.; Sussan, T.E.; Chen, M.; Dobzanski, A.; Lane, A.P.; Sidhaye, V.; Biswal, S.; Ramanathan, M.J. Deletion of Nrf2 Enhances Susceptibility to Eosinophilic Sinonasal Inflammation in a Murine Model of Rhinosinusitis. Int. Forum Allergy Rhinol. 2019, 9, 114–119.
- London, N.R.J.; Tharakan, A.; Lane, A.P.; Biswal, S.; Ramanathan, M.J. Nuclear Erythroid 2-Related Factor 2 Activation Inhibits House Dust Mite-Induced Sinonasal Epithelial Cell Barrier Dysfunction. Int. Forum Allergy Rhinol. 2017, 7, 536–541.
- Nishida, M.; Takeno, S.; Takemoto, K.; Takahara, D.; Hamamoto, T.; Ishino, T.; Kawasumi, T. Increased Tissue Expression of Lectin-Like Oxidized LDL Receptor-1 (LOX-1) Is Associated with Disease Severity in Chronic Rhinosinusitis with Nasal Polyps. Diagnostics 2020, 10, 246.
- Kawasumi, T.; Takeno, S.; Ishikawa, C.; Takahara, D.; Taruya, T.; Takemoto, K.; Hamamoto, T.; Ishino, T.; Ueda, T. The Functional Diversity of Nitric Oxide Synthase Isoforms in Human Nose and Paranasal Sinuses: Contrasting Pathophysiological Aspects in Nasal Allergy and Chronic Rhinosinusitis. Int. J. Mol. Sci. 2021, 22, 7561.
- Paoletti, G.; Casini, M.; Malvezzi, L.; Pirola, F.; Russo, E.; Nappi, E.; Quintina Muci, G.; Montagna, C.; Messina, M.R.; Ferri, S.; et al. Very Rapid Improvement of Extended Nitric Oxide Parameters, Associated with Clinical and Functional Betterment, in Patients with Chronic Rhinosinusitis with Nasal Polyps (CRSwNP) Treated with Dupilumab. J. Investig. Allergol. Clin. Immunol. 2022, 33, 1–25.
- Jeruzal-Świątecka, J.; Borkowska, E.; Łaszczych, M.; Nowicka, Z.; Pietruszewska, W. TAS2R38 Bitter Taste Receptor Expression in Chronic Rhinosinusitis with Nasal Polyps: New Data on Polypoid Tissue. Int. J. Mol. Sci. 2022, 23, 7345.
- Cantone, E.; Negri, R.; Roscetto, E.; Grassia, R.; Catania, M.R.; Capasso, P.; Maffei, M.; Soriano, A.A.; Leone, C.A.; Iengo, M.; et al. In Vivo Biofilm Formation, Gram-Negative Infections and TAS2R38 Polymorphisms in CRSw NP Patients. Laryngoscope 2018, 128, E339–E345.
- Carey, R.M.; Hariri, B.M.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages. Cells 2022, 11, 1478.
- Workman, A.D.; Maina, I.W.; Brooks, S.G.; Kohanski, M.A.; Cowart, B.J.; Mansfield, C.; Kennedy, D.W.; Palmer, J.N.; Adappa, N.D.; Reed, D.R.; et al. The Role of Quinine-Responsive Taste Receptor Family 2 in Airway Immune Defense and Chronic Rhinosinusitis. Front. Immunol. 2018, 9, 624.
- Qu, J.; Mei, Q.; Niu, R. Oxidative CaMKII as a Potential Target for Inflammatory Disease (Review). Mol. Med. Rep. 2019, 20, 863–870.
- Wang, H.; Do, D.C.; Liu, J.; Wang, B.; Qu, J.; Ke, X.; Luo, X.; Tang, H.M.; Tang, H.L.; Hu, C.; et al. Functional Role of Kynurenine and Aryl Hydrocarbon Receptor Axis in Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. 2018, 141, 586–600.
- Esen, E.; Selçuk, A.; Passali, D. Epidemiology of Nasal Polyposis. In All Around the Nose; Cingi, C., Bayar Muluk, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 367–371. ISBN 978-3-030-21217-9.
- Rajguru, R. Nasal Polyposis: Current Trends. Indian J. Otolaryngol. Head Neck Surg. 2014, 66, 16–21.
- Traina, G.; Bolzacchini, E.; Bonini, M.; Contini, D.; Mantecca, P.; Caimmi, S.M.E.; Licari, A. Role of Air Pollutants Mediated Oxidative Stress in Respiratory Diseases. Pediatr. Allergy Immunol. 2022, 33 (Suppl. S27), 38–40.
- Istratenco, A. Oxidative Stress-Related Pathophysiology in Chronic Rhinosinusitis with Nasal Polyps: Research Challenges. Rom. J. Rhinol. 2019, 9, 71–77.
- Cho, D.-Y.; Le, W.; Bravo, D.T.; Hwang, P.H.; Illek, B.; Fischer, H.; Nayak, J. V Air Pollutants Cause Release of Hydrogen Peroxide and Interleukin-8 in a Human Primary Nasal Tissue Culture Model. Int. Forum Allergy Rhinol. 2014, 4, 966–971.
- Holecek, V.; Rokyta, R.; Slipka, J. Free Radicals in Nasal and Paranasal Diseases. In Free Radicals in ENT Pathology; Miller, J., Le Prell, C.G., Rybak, L., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 479–492. ISBN 978-3-319-13473-4.
- Topal, O.; Kulaksızoglu, S.; Erbek, S.S. Oxidative Stress and Nasal Polyposis: Does It Affect the Severity of the Disease? Am. J. Rhinol. Allergy 2014, 28, e1–e4.
- Okur, E.; Gul, A.; Kilinc, M.; Kilic, M.A.; Yildirim, I.; Tolun, F.I.; Atli, Y. Trace Elements in Nasal Polyps. Eur. Arch. Oto-Rhino-Laryngol. 2013, 270, 2245–2248.
- Cheng, Y.-K.; Hwang, G.-Y.; Lin, C.-D.; Tsai, M.-H.; Tsai, S.-W.; Chang, W.-C. Altered Expression Profile of Superoxide Dismutase Isoforms in Nasal Polyps from Nonallergic Patients. Laryngoscope 2006, 116, 417–422.
- Cekin, E.; Ipcioglu, O.M.; Erkul, B.E.; Kapucu, B.; Ozcan, O.; Cincik, H.; Gungor, A. The Association of Oxidative Stress and Nasal Polyposis. J. Int. Med. Res. 2009, 37, 325–330.
- Sagit, M.; Erdamar, H.; Saka, C.; Yalcin, S.; Akin, I. Effect of Antioxidants on the Clinical Outcome of Patients with Nasal Polyposis. J. Laryngol. Otol. 2011, 125, 811–815.
- Simsek, F.; Eren, E.; Bahceci, S.; Aladag, I. High PI3K/MTOR and Low MAPK/JNK Activity Results in Decreased Apoptosis and Autophagy in Nasal Polyposis. Braz. J. Otorhinolaryngol. 2021, 87, 572–577.
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, Ö. Oxidative Stress in Asthma: Part of the Puzzle. Pediatr. Allergy Immunol. 2018, 29, 789–800.
- Okeleji, L.O.; Ajayi, A.F.; Adebayo-Gege, G.; Aremu, V.O.; Adebayo, O.I.; Adebayo, E.T. Epidemiologic Evidence Linking Oxidative Stress and Pulmonary Function in Healthy Populations. Chronic Dis. Transl. Med. 2021, 7, 88–99.
- Ricciardolo, F.L.M.; Bertolini, F.; Carriero, V.; Sprio, A.E. Asthma Phenotypes and Endotypes. Minerva Med. 2021, 112, 547–563.
- Kleniewska, P.; Pawliczak, R. The Participation of Oxidative Stress in the Pathogenesis of Bronchial Asthma. Biomed. Pharmacother. 2017, 94, 100–108.
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233.
- Gans, M.D.; Gavrilova, T. Understanding the Immunology of Asthma: Pathophysiology, Biomarkers, and Treatments for Asthma Endotypes. Paediatr. Respir. Rev. 2020, 36, 118–127.
- Jones, T.L.; Neville, D.M.; Chauhan, A.J. Diagnosis and Treatment of Severe Asthma: A Phenotype-Based Approach. Clin. Med. 2018, 18, s36–s40.
- Katial, R.K.; Bensch, G.W.; Busse, W.W.; Chipps, B.E.; Denson, J.L.; Gerber, A.N.; Jacobs, J.S.; Kraft, M.; Martin, R.J.; Nair, P.; et al. Changing Paradigms in the Treatment of Severe Asthma: The Role of Biologic Therapies. J. Allergy Clin. Immunol. Pract. 2017, 5, S1–S14.
- Chamitava, L.; Cazzoletti, L.; Ferrari, M.; Garcia-Larsen, V.; Jalil, A.; Degan, P.; Fois, A.G.; Zinellu, E.; Fois, S.S.; Fratta Pasini, A.M.; et al. Biomarkers of Oxidative Stress and Inflammation in Chronic Airway Diseases. Int. J. Mol. Sci. 2020, 21, 4339.
- Allam, V.S.R.R.; Paudel, K.R.; Gupta, G.; Singh, S.K.; Vishwas, S.; Gulati, M.; Gupta, S.; Chaitanya, M.V.N.L.; Jha, N.K.; Gupta, P.K.; et al. Nutraceuticals and Mitochondrial Oxidative Stress: Bridging the Gap in the Management of Bronchial Asthma. Environ. Sci. Pollut. Res. 2022, 29, 62733–62754.
- Liu, K.; Hua, S.; Song, L. PM2.5 Exposure and Asthma Development: The Key Role of Oxidative Stress. Oxid. Med. Cell. Longev. 2022, 2022, 3618806.
- Tiotiu, A.I.; Novakova, P.; Nedeva, D.; Chong-Neto, H.J.; Novakova, S.; Steiropoulos, P.; Kowal, K. Impact of Air Pollution on Asthma Outcomes. Int. J. Environ. Res. Public Health 2020, 17, 6212.
- Jesenak, M.; Zelieskova, M.; Babusikova, E. Oxidative Stress and Bronchial Asthma in Children—Causes or Consequences? Front. Pediatr. 2017, 5, 162.
- Xu, C.; Song, Y.; Wang, Z.; Jiang, J.; Piao, Y.; Li, L.; Jin, S.; Li, L.; Zhu, L.; Yan, G. Pterostilbene Suppresses Oxidative Stress and Allergic Airway Inflammation through AMPK/Sirt1 and Nrf2/HO-1 Pathways. Immunity, Inflamm. Dis. 2021, 9, 1406–1417.
- Yong, J.I.; Kim, D.W.; Shin, M.J.; Jo, H.S.; Park, J.H.; Cho, S.B.; Lee, C.H.; Yeo, H.J.; Yeo, E.J.; Choi, Y.J.; et al. PEP-1-PEA15 Suppresses Inflammatory Responses by Regulation of MAPK in Macrophages and Animal Models. Immunobiology 2018, 223, 709–717.
- Zhang, X.; Ding, M.; Zhu, P.; Huang, H.; Zhuang, Q.; Shen, J.; Cai, Y.; Zhao, M.; He, Q. New Insights into the Nrf-2/HO-1 Signaling Axis and Its Application in Pediatric Respiratory Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3214196.
- Li, J.; Wang, H.; Zheng, Z.; Luo, L.; Wang, P.; Liu, K.; Namani, A.; Jiang, Z.; Wang, X.J.; Tang, X. Mkp-1 Cross-Talks with Nrf2/Ho-1 Pathway Protecting against Intestinal Inflammation. Free Radic. Biol. Med. 2018, 124, 541–549.
- Wang, C.; Choi, Y.H.; Xian, Z.; Zheng, M.; Piao, H.; Yan, G. Aloperine Suppresses Allergic Airway Inflammation through NF-ΚB, MAPK, and Nrf2/HO-1 Signaling Pathways in Mice. Int. Immunopharmacol. 2018, 65, 571–579.
- Gu, X.; Zhang, Q.; Du, Q.; Shen, H.; Zhu, Z. Pinocembrin Attenuates Allergic Airway Inflammation via Inhibition of NF-ΚB Pathway in Mice. Int. Immunopharmacol. 2017, 53, 90–95.
- Yi, L.; Cui, J.; Wang, W.; Tang, W.; Teng, F.; Zhu, X.; Qin, J.; Wuniqiemu, T.; Sun, J.; Wei, Y.; et al. Formononetin Attenuates Airway Inflammation and Oxidative Stress in Murine Allergic Asthma. Front. Pharmacol. 2020, 11, 533841.
- Lim, J.-O.; Song, K.H.; Lee, I.S.; Lee, S.-J.; Kim, W.-I.; Pak, S.-W.; Shin, I.-S.; Kim, T. Cimicifugae Rhizoma Extract Attenuates Oxidative Stress and Airway Inflammation via the Upregulation of Nrf2/HO-1/NQO1 and Downregulation of NF-ΚB Phosphorylation in Ovalbumin-Induced Asthma. Antioxidants 2021, 10, 1626.
- Suzuki, M.; Cole, J.J.; Konno, S.; Makita, H.; Kimura, H.; Nishimura, M.; Maciewicz, R.A. Large-Scale Plasma Proteomics Can Reveal Distinct Endotypes in Chronic Obstructive Pulmonary Disease and Severe Asthma. Clin. Transl. Allergy 2021, 11, e12091.
- Saunders, R.M.; Biddle, M.; Amrani, Y.; Brightling, C.E. Stressed out—The Role of Oxidative Stress in Airway Smooth Muscle Dysfunction in Asthma and COPD. Free Radic. Biol. Med. 2022, 185, 97–119.
- Carpagnano, G.E.; Scioscia, G.; Lacedonia, D.; Soccio, P.; Quarato, C.M.I.; Cotugno, G.; Palumbo, M.G.; Foschino Barbaro, M.P. Searching for Inflammatory and Oxidative Stress Markers Capable of Clustering Severe Asthma. Arch. Bronconeumol. 2021, 57, 338–344.
- Lewis, B.W.; Ford, M.L.; Rogers, L.K.; Britt, R.D. Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD. Antioxidants 2021, 10, 1335.
- Enweasor, C.; Flayer, C.H.; Haczku, A. Ozone-Induced Oxidative Stress, Neutrophilic Airway Inflammation, and Glucocorticoid Resistance in Asthma. Front. Immunol. 2021, 12, 631092.

